
Hi I have a set of tetramers created by AlphaFold. They are randomly oriented. In order to measure their pore size in a third-party program, I need to orient them with their z axis pointing in the z direction. I can orient them with respect to the screen, but I don't know how to save this view orientation (i.e. so that the z axis in the pdb file goes vertically). Is there any way how to do it? The switch in the "save" command specifying whether to use transformed or untransformed coordinates does not make any difference, and the file is always saved in the original orientation. Best Sasha

Hi Sasha, When you just rotate the whole view, the model is not transformed, only the point of view. To actually transform the model position, you'd have move it (or them, if multiple) using some model-specific mouse mode, not the usual mousemodes that move the whole view. At least according to my tests, if you move all the models in sync, it will always move the whole view instead of transforming the individual models. Soe what you could do is (1) open the structure(s) of interest and then open some other structure (doesn't matter what, you will ignore the latter, it's just there as an anchor (2) DO NOT move the scene as a whole, i.e. absolutely no rotation or translation after you open the models (3) use a model-specific mousemode to rotate only the model(s) of interest and leave the other unmoved - for example, in the Right Mouse tab of icons across the top, choose "rotate selected models," (icon looks like selected water molecule with curved arrows around it), Ctrl-click to select some part of your model of interest (if your tetramer is 4 separate models, you would have to select at least 1 atom in each), then use right mouse to rotate it/them. See Right Mouse help: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/mousemodes.html> (4) then save your model(s) "relative" to the other model that you didn't move I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 29, 2025, at 4:46 AM, Alexandra Zahradnikova via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi I have a set of tetramers created by AlphaFold. They are randomly oriented. In order to measure their pore size in a third-party program, I need to orient them with their z axis pointing in the z direction. I can orient them with respect to the screen, but I don't know how to save this view orientation (i.e. so that the z axis in the pdb file goes vertically). Is there any way how to do it? The switch in the "save" command specifying whether to use transformed or untransformed coordinates does not make any difference, and the file is always saved in the original orientation. Best Sasha
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/

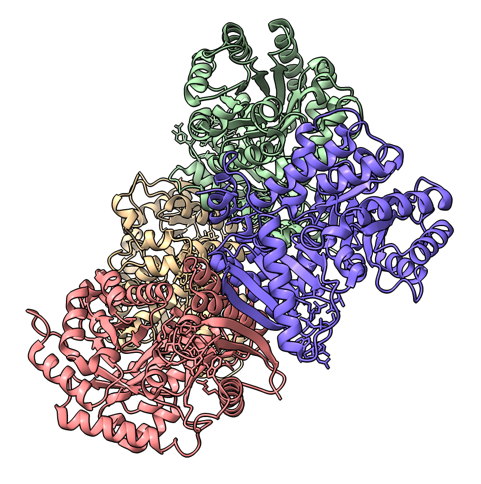
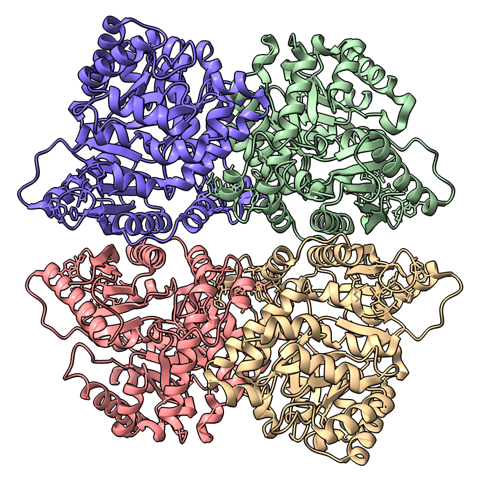
Hi Sasha, Here's another idea about how to align a tetramer with symmetry axis along z and save it. The idea is to create 4 atoms in the aligned arrangement using the ChimeraX sym command, and then align the tetramer model to those 4 atoms. Here's an example using PDB 9r3i which is a tetramer with D2 symmetry. open 9r3i del ~/A:94@CA sym #1 D2 hide ribbon ; show atoms ; style sphere ; view orient open 9r3i color bychain align #3/A,B,D,C:94@CA to #2.1,3,2,4 save aligned_9r3i.cif model #3 I chose the CA atom of residue 94 chain A arbitrarily just picking one on the periphery of the tetramer. The align command is a bit tricky. I had to put the chains A,B,C,D and symmetric 4 atoms I made in the same clockwise order. Mousing over the 4 chains of 9r3i to see the popup window with the chain names shows they are in order A,B,D,C as I go clockwise, and my 4 atoms placed with D2 symmetry are in clockwise order #2.1, #2.3, #2.2, #2.4. So I used these orders in the align command. If you make tetramer predictions with AlphaFold, the 4 chains will ikely end up in different orders for different predictions. If you are comparing tetramers I would align one along z with the above procedure and then align each of the others to this one by choosing 4 atoms in each and using the align command. Tom PDB 9r3i original and z aligned.
On Sep 29, 2025, at 10:10 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Sasha, When you just rotate the whole view, the model is not transformed, only the point of view. To actually transform the model position, you'd have move it (or them, if multiple) using some model-specific mouse mode, not the usual mousemodes that move the whole view. At least according to my tests, if you move all the models in sync, it will always move the whole view instead of transforming the individual models.
Soe what you could do is
(1) open the structure(s) of interest and then open some other structure (doesn't matter what, you will ignore the latter, it's just there as an anchor
(2) DO NOT move the scene as a whole, i.e. absolutely no rotation or translation after you open the models
(3) use a model-specific mousemode to rotate only the model(s) of interest and leave the other unmoved
- for example, in the Right Mouse tab of icons across the top, choose "rotate selected models," (icon looks like selected water molecule with curved arrows around it), Ctrl-click to select some part of your model of interest (if your tetramer is 4 separate models, you would have to select at least 1 atom in each), then use right mouse to rotate it/them. See Right Mouse help: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/mousemodes.html>
(4) then save your model(s) "relative" to the other model that you didn't move
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 29, 2025, at 4:46 AM, Alexandra Zahradnikova via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi I have a set of tetramers created by AlphaFold. They are randomly oriented. In order to measure their pore size in a third-party program, I need to orient them with their z axis pointing in the z direction. I can orient them with respect to the screen, but I don't know how to save this view orientation (i.e. so that the z axis in the pdb file goes vertically). Is there any way how to do it? The switch in the "save" command specifying whether to use transformed or untransformed coordinates does not make any difference, and the file is always saved in the original orientation. Best Sasha
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
{kind=link}
Thanks Elaine Best Sasha -----Original Message----- From: Elaine Meng [mailto:meng@cgl.ucsf.edu] Sent: Monday, September 29, 2025 19:10 To: Alexandra Zahradnikova Cc: ChimeraX Users Help Subject: Re: [chimerax-users] Orient many tetramers Hi Sasha, When you just rotate the whole view, the model is not transformed, only the point of view. To actually transform the model position, you'd have move it (or them, if multiple) using some model-specific mouse mode, not the usual mousemodes that move the whole view. At least according to my tests, if you move all the models in sync, it will always move the whole view instead of transforming the individual models. Soe what you could do is (1) open the structure(s) of interest and then open some other structure (doesn't matter what, you will ignore the latter, it's just there as an anchor (2) DO NOT move the scene as a whole, i.e. absolutely no rotation or translation after you open the models (3) use a model-specific mousemode to rotate only the model(s) of interest and leave the other unmoved - for example, in the Right Mouse tab of icons across the top, choose "rotate selected models," (icon looks like selected water molecule with curved arrows around it), Ctrl-click to select some part of your model of interest (if your tetramer is 4 separate models, you would have to select at least 1 atom in each), then use right mouse to rotate it/them. See Right Mouse help: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/mousemodes.html> (4) then save your model(s) "relative" to the other model that you didn't move I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 29, 2025, at 4:46 AM, Alexandra Zahradnikova via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi I have a set of tetramers created by AlphaFold. They are randomly oriented. In order to measure their pore size in a third-party program, I need to orient them with their z axis pointing in the z direction. I can orient them with respect to the screen, but I don't know how to save this view orientation (i.e. so that the z axis in the pdb file goes vertically). Is there any way how to do it? The switch in the "save" command specifying whether to use transformed or untransformed coordinates does not make any difference, and the file is always saved in the original orientation. Best Sasha
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hi Tom Many thanks! Actually, since my channels have a C4 symmetry, they are correctly aligned with respect to the z axis even if the numbering of chains is random (I have tried 5 variants with different chain sequence). This simplifies things a lot. Best Sasha -----Original Message----- From: Tom Goddard [mailto:goddard@sonic.net] Sent: Monday, September 29, 2025 20:39 To: Alexandra Zahradnikova Cc: ChimeraX Users Help Subject: Re: [chimerax-users] Orient many tetramers Hi Sasha, Here's another idea about how to align a tetramer with symmetry axis along z and save it. The idea is to create 4 atoms in the aligned arrangement using the ChimeraX sym command, and then align the tetramer model to those 4 atoms. Here's an example using PDB 9r3i which is a tetramer with D2 symmetry. open 9r3i del ~/A:94@CA sym #1 D2 hide ribbon ; show atoms ; style sphere ; view orient open 9r3i color bychain align #3/A,B,D,C:94@CA to #2.1,3,2,4 save aligned_9r3i.cif model #3 I chose the CA atom of residue 94 chain A arbitrarily just picking one on the periphery of the tetramer. The align command is a bit tricky. I had to put the chains A,B,C,D and symmetric 4 atoms I made in the same clockwise order. Mousing over the 4 chains of 9r3i to see the popup window with the chain names shows they are in order A,B,D,C as I go clockwise, and my 4 atoms placed with D2 symmetry are in clockwise order #2.1, #2.3, #2.2, #2.4. So I used these orders in the align command. If you make tetramer predictions with AlphaFold, the 4 chains will ikely end up in different orders for different predictions. If you are comparing tetramers I would align one along z with the above procedure and then align each of the others to this one by choosing 4 atoms in each and using the align command. Tom PDB 9r3i original and z aligned.
participants (3)
-
 Alexandra Zahradnikova
Alexandra Zahradnikova -
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard