Changing range for electrostatic potential coloring

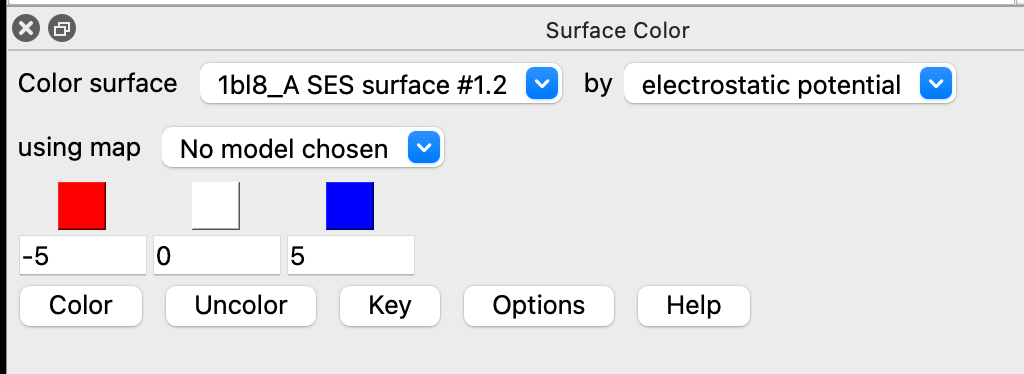
I want to have less white and more color on an electrostatic potential map. I suspect this is done by reducing the range, but I can't find how to do this. I start with open 1bl8 fromDatabase pdb format mmcif select protein coulombic sel ui tool show "Surface Color" Then I have this dialog, where I have changed -10 ... +10 to -5 ... +5 When I click the Color button, I get "No map chosen for coloring", but the "using map" menu has no options. ??? Also I would like to re-color the surfaces for all 4 chains, not one at a time, but the "Color surface" menu seems to have 4 separate chain surfaces, but no option for all 4 at once. I will appreciate guidance! Thanks, -Erichttp://martz.molviz.org
{kind=link}

Hi Eric, The coulombic comand does not produce a map of electrostatic potential by default, so the Surface Color that colors a surface using map values will not be of use. Instead you can use the coulombic command "range" option, for example coulombic sel range -5,5 This is explained in the coulombic command documentation with an example here https://www.cgl.ucsf.edu/chimerax/docs/user/commands/coulombic.html#palette-... Tom
On Dec 5, 2024, at 2:35 PM, Eric Martz via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
I want to have less white and more color on an electrostatic potential map. I suspect this is done by reducing the range, but I can't find how to do this. I start with
open 1bl8 fromDatabase pdb format mmcif select protein coulombic sel ui tool show "Surface Color"
Then I have this dialog, where I have changed -10 ... +10 to -5 ... +5
<Screenshot 2024-12-05 at 5.27.57 PM.png>
When I click the Color button, I get "No map chosen for coloring", but the "using map" menu has no options.
???
Also I would like to re-color the surfaces for all 4 chains, not one at a time, but the "Color surface" menu seems to have 4 separate chain surfaces, but no option for all 4 at once.
I will appreciate guidance!
Thanks, -Eric http://martz.molviz.org <http://martz.molviz.org/>
<Screenshot 2024-12-05 at 5.27.57 PM.png>_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Wonderful! Thanks, Tom! -Eric On Thursday, December 5, 2024 at 05:55:33 PM EST, Tom Goddard <goddard@sonic.net> wrote: Hi Eric, The coulombic comand does not produce a map of electrostatic potential by default, so the Surface Color that colors a surface using map values will not be of use. Instead you can use the coulombic command "range" option, for example coulombic sel range -5,5 This is explained in the coulombic command documentation with an example here https://www.cgl.ucsf.edu/chimerax/docs/user/commands/coulombic.html#palette-... Tom On Dec 5, 2024, at 2:35 PM, Eric Martz via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: I want to have less white and more color on an electrostatic potential map. I suspect this is done by reducing the range, but I can't find how to do this. I start with open 1bl8 fromDatabase pdb format mmcif select protein coulombic sel ui tool show "Surface Color" Then I have this dialog, where I have changed -10 ... +10 to -5 ... +5 <Screenshot 2024-12-05 at 5.27.57 PM.png> When I click the Color button, I get "No map chosen for coloring", but the "using map" menu has no options. ??? Also I would like to re-color the surfaces for all 4 chains, not one at a time, but the "Color surface" menu seems to have 4 separate chain surfaces, but no option for all 4 at once. I will appreciate guidance! Thanks, -Erichttp://martz.molviz.org <Screenshot 2024-12-05 at 5.27.57 PM.png>_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
You may be interested in seeing this comparison of electrostatic potential maps generated by ChimeraX, iCn3D, and PyMOL: https://proteopedia.org/wiki/index.php/Electrostatic_potential_maps and the related topic https://proteopedia.org/wiki/index.php/Missing_residues_and_incomplete_sidec... suggests using AlphaFold models for electrostatic potential maps when the empirical model lacks sidechains of charged amino acids on its surface (very common). -Eric Martzhttp://martz.molviz.org

Thanks, Eric! It's good to bring people's attention to the problem of missing atoms when calculating electrostatic potential. I think we have some warnings in the Log when there are missing parts, but probably many people disregard them. Besides using AlphaFold, Chimera and ChimeraX also have a Dock Prep tool that can fill in truncated amino acid sidechains using a rotamer library, and a Modeller interface to fill in missing loops where the backbone is also missing. AlphaFold may do a better job on the exact placement, however; I have not done a detailed comparison. ChimeraX can also use a potential map from APBS, DelPhi, etc. for coloring, but the built-in calculation is just the simple Coulombic one. A LONG time ago I made a page informally discussing surface coloring with simple Coulombic ESP vs. maps calculated with the full Poisson-Boltzmann approach: <https://www.cgl.ucsf.edu/home/meng/grpmt/esp-compare/esp-compare.html> Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 6, 2024, at 1:52 PM, Eric Martz via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
You may be interested in seeing this comparison of electrostatic potential maps generated by ChimeraX, iCn3D, and PyMOL:
https://proteopedia.org/wiki/index.php/Electrostatic_potential_maps and the related topic
https://proteopedia.org/wiki/index.php/Missing_residues_and_incomplete_sidec... which suggests using AlphaFold models for electrostatic potential maps when the empirical model lacks sidechains of charged amino acids on its surface (very common).
-Eric Martz http://martz.molviz.org _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Dear Elaine, Dock Prep: Very nice! I ran it on 1bl8 and confirmed that it filled in missing sidechain atoms on 20 residues, and added hydrogens. I'll add mention of it in my articles linked below. Thanks for calling it to my attention! Also I appreciate your 2009 Coulombic coloring comparison. I'll also link that. -Eric On Friday, December 6, 2024 at 09:23:19 PM EST, Elaine Meng <meng@cgl.ucsf.edu> wrote: Thanks, Eric! It's good to bring people's attention to the problem of missing atoms when calculating electrostatic potential. I think we have some warnings in the Log when there are missing parts, but probably many people disregard them. Besides using AlphaFold, Chimera and ChimeraX also have a Dock Prep tool that can fill in truncated amino acid sidechains using a rotamer library, and a Modeller interface to fill in missing loops where the backbone is also missing. AlphaFold may do a better job on the exact placement, however; I have not done a detailed comparison. ChimeraX can also use a potential map from APBS, DelPhi, etc. for coloring, but the built-in calculation is just the simple Coulombic one. A LONG time ago I made a page informally discussing surface coloring with simple Coulombic ESP vs. maps calculated with the full Poisson-Boltzmann approach: <https://www.cgl.ucsf.edu/home/meng/grpmt/esp-compare/esp-compare.html> Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 6, 2024, at 1:52 PM, Eric Martz via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
You may be interested in seeing this comparison of electrostatic potential maps generated by ChimeraX, iCn3D, and PyMOL:
https://proteopedia.org/wiki/index.php/Electrostatic_potential_maps and the related topic
https://proteopedia.org/wiki/index.php/Missing_residues_and_incomplete_sidec... which suggests using AlphaFold models for electrostatic potential maps when the empirical model lacks sidechains of charged amino acids on its surface (very common).
-Eric Martz http://martz.molviz.org _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
participants (3)
-
 Elaine Meng
Elaine Meng -
 Eric Martz
Eric Martz -
 Tom Goddard
Tom Goddard