RMSD does not make sense in a few cases

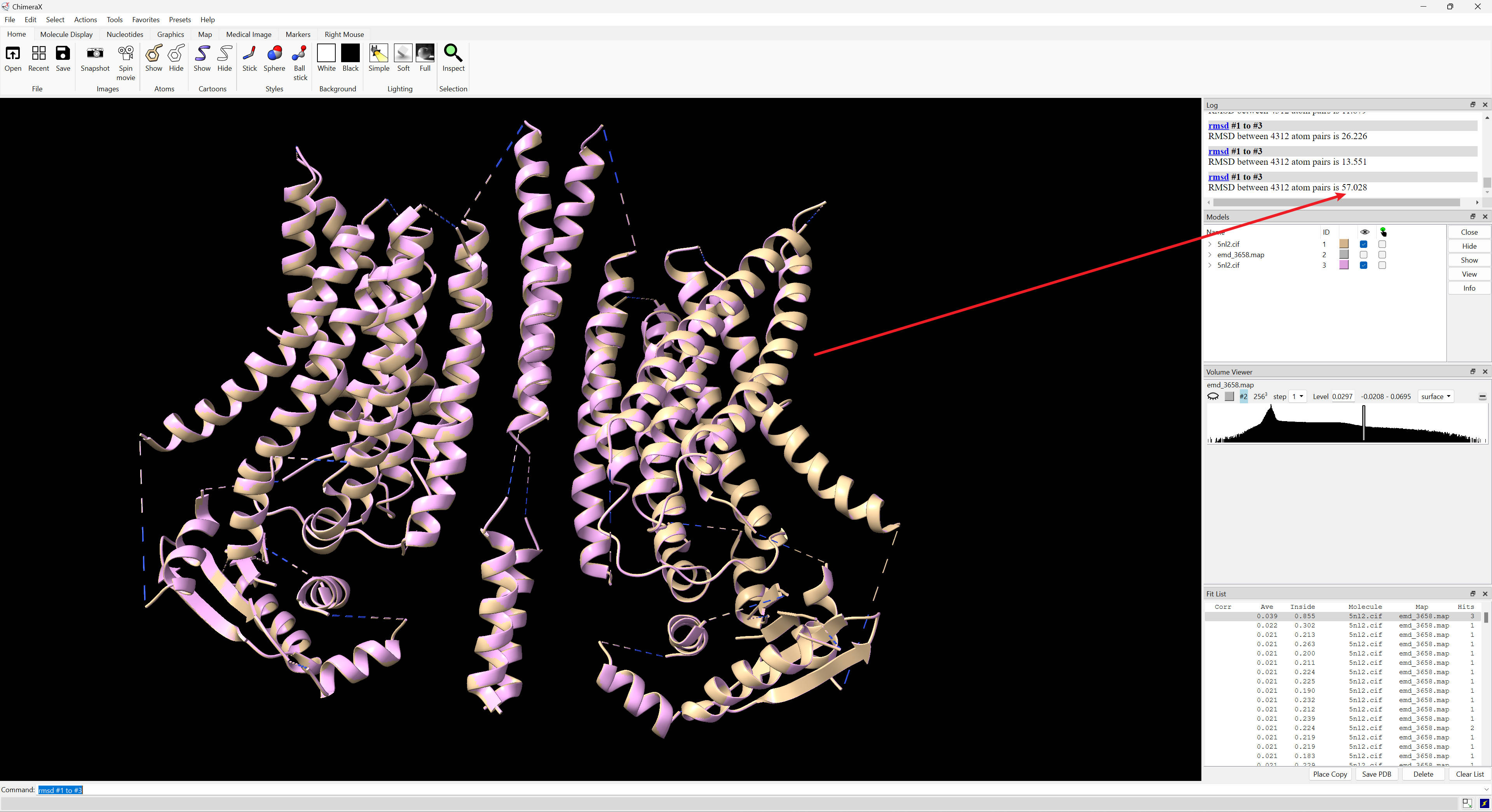
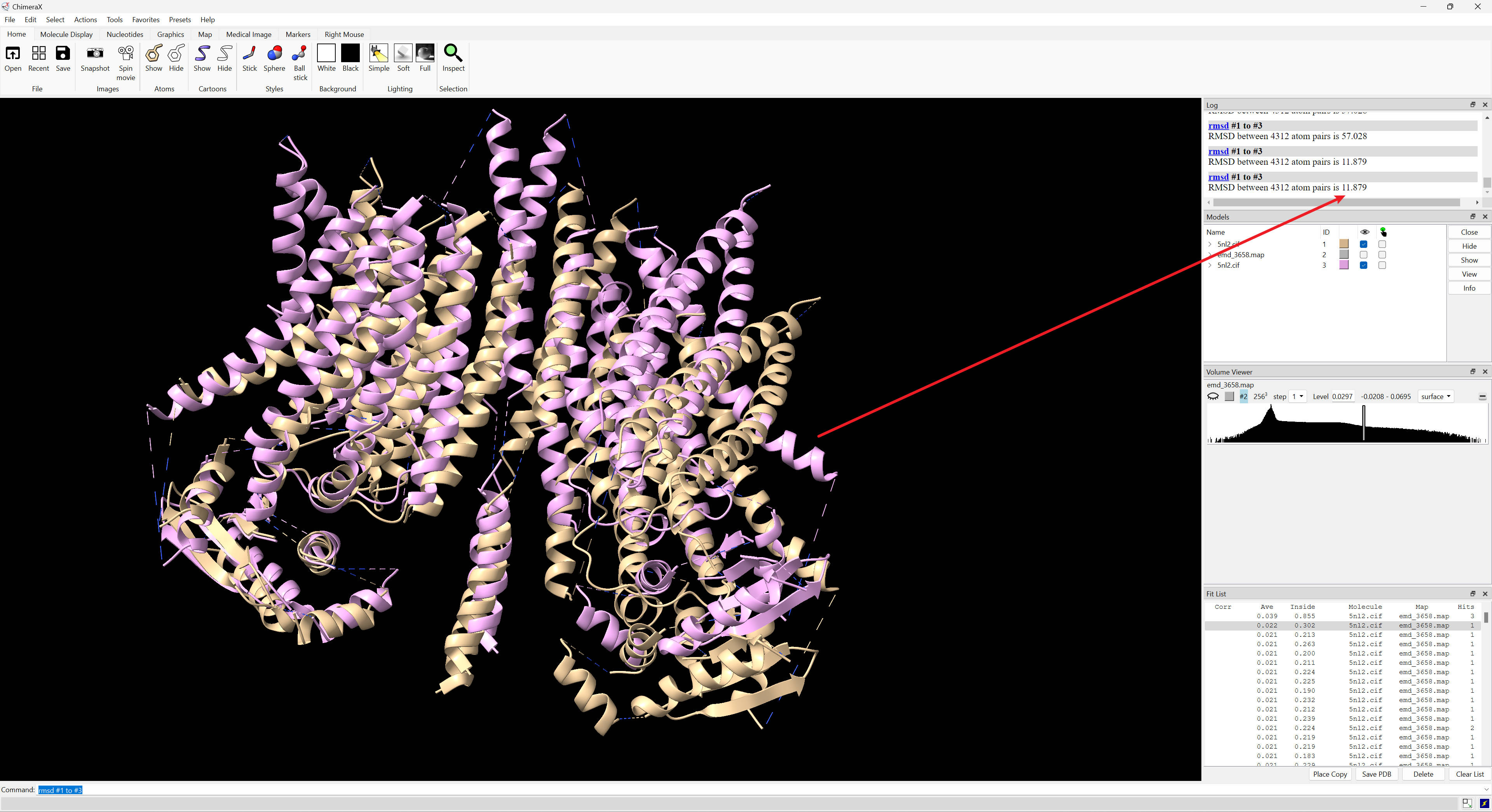
Hello ChimeraX Team, In the attached screenshot, #1 and #3 are almost overlapping, but get an RMSD as 57. I occasionally encounter this. The session and log files are attached. In short, to reproduce, open 5nl2.cif and emd_3658.map; fitmap #1 in #2 search 1000; rmsd #1 to #3. This happened rarely, but in this case, it happened twice. It might be that I fit other structures first. But I indeed click "Clear List" and "close session" after each fitting. The second top hit's RMSD seems to make sense in the attached session (screenshot attached too). Thanks, Roden -- This message and its contents, including attachments are intended solely for the original recipient. If you are not the intended recipient or have received this message in error, please notify me immediately and delete this message from your computer system. Any unauthorized use or distribution is prohibited. Please consider the environment before printing this email.
{kind=link}
{kind=link}

Hi Roden, PDB 5nl2 is symmetric and when you fit it to a map and it is flipped 180 degrees you get an RMSD of 57 Angstroms. Tom
On Jun 28, 2024, at 11:59 PM, Roden Deng Luo via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello ChimeraX Team,
In the attached screenshot, #1 and #3 are almost overlapping, but get an RMSD as 57. I occasionally encounter this. The session and log files are attached. In short, to reproduce, open 5nl2.cif and emd_3658.map; fitmap #1 in #2 search 1000; rmsd #1 to #3. This happened rarely, but in this case, it happened twice. It might be that I fit other structures first. But I indeed click "Clear List" and "close session" after each fitting. The second top hit's RMSD seems to make sense in the attached session (screenshot attached too).
Thanks, Roden
--
This message and its contents, including attachments are intended solely for the original recipient. If you are not the intended recipient or have received this message in error, please notify me immediately and delete this message from your computer system. Any unauthorized use or distribution is prohibited. Please consider the environment before printing this email. <RMSD error.txt><RMSD bug.jpg><RMSD error.cxs><2nd fit RMSD.jpg>_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Aha! Thanks, Tom! I will then use the following command to turn the molecule and calculate a working RMSD in such cases. Kindly correct me if I'm wrong. turn z 1 180 model #1 center #1 coordinateSystem #1 Best, Roden On Sat, Jun 29, 2024 at 7:01 PM Tom Goddard <goddard@sonic.net> wrote:
Hi Roden,
PDB 5nl2 is symmetric and when you fit it to a map and it is flipped 180 degrees you get an RMSD of 57 Angstroms.
Tom
On Jun 28, 2024, at 11:59 PM, Roden Deng Luo via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello ChimeraX Team,
In the attached screenshot, #1 and #3 are almost overlapping, but get an RMSD as 57. I occasionally encounter this. The session and log files are attached. In short, to reproduce, open 5nl2.cif and emd_3658.map; fitmap #1 in #2 search 1000; rmsd #1 to #3. This happened rarely, but in this case, it happened twice. It might be that I fit other structures first. But I indeed click "Clear List" and "close session" after each fitting. The second top hit's RMSD seems to make sense in the attached session (screenshot attached too).
Thanks, Roden
--
This message and its contents, including attachments are intended solely for the original recipient. If you are not the intended recipient or have received this message in error, please notify me immediately and delete this message from your computer system. Any unauthorized use or distribution is prohibited. Please consider the environment before printing this email. <RMSD error.txt><RMSD bug.jpg><RMSD error.cxs><2nd fit RMSD.jpg>_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://urldefense.com/v3/__https://mail.cgl.ucsf.edu/mailman/archives/list/...
-- This message and its contents, including attachments are intended solely for the original recipient. If you are not the intended recipient or have received this message in error, please notify me immediately and delete this message from your computer system. Any unauthorized use or distribution is prohibited. Please consider the environment before printing this email.

Hey Roden, If you color by chain, you'll see the issue. I think the rmsd you're getting is comparing chain A to chain A and chain B to chain B. Since A is aligned to B, the reported RMSD won't make sense. I guess you could rename the chains, or just use matchmaker to align them (mmaker #1 to #3). Here's the description of rmsd from "help rmsd": The two sets must contain the same number of atoms, which will be paired in the order given. If the order is not given explicitly, chains within a model are sorted by ID, residues with a chain sorted by number, and atoms within a residue sorted by name before pairing. Harper ________________________________ From: Roden Deng Luo via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Sent: Saturday, June 29, 2024 2:59 AM To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: [chimerax-users] RMSD does not make sense in a few cases Hello ChimeraX Team, In the attached screenshot, #1 and #3 are almost overlapping, but get an RMSD as 57. I occasionally encounter this. The session and log files are attached. In short, to reproduce, open 5nl2.cif and emd_3658.map; fitmap #1 in #2 search 1000; rmsd #1 to #3. This happened rarely, but in this case, it happened twice. It might be that I fit other structures first. But I indeed click "Clear List" and "close session" after each fitting. The second top hit's RMSD seems to make sense in the attached session (screenshot attached too). Thanks, Roden -- This message and its contents, including attachments are intended solely for the original recipient. If you are not the intended recipient or have received this message in error, please notify me immediately and delete this message from your computer system. Any unauthorized use or distribution is prohibited. Please consider the environment before printing this email.
Hi Harper, Many thanks! After applying the following command for this specific case, the RMSD does make sense now. changechains #1 A,B B,A Cheers, Roden On Sat, Jun 29, 2024 at 8:30 PM Smith, Harper <smith.12510@buckeyemail.osu.edu> wrote:
Hey Roden,
If you color by chain, you'll see the issue. I think the rmsd you're getting is comparing chain A to chain A and chain B to chain B. Since A is aligned to B, the reported RMSD won't make sense. I guess you could rename the chains, or just use matchmaker to align them (mmaker #1 to #3).
Here's the description of rmsd from "help rmsd":
The two sets must contain the same number of atoms, which will be paired in the order given. If the order is not given explicitly, chains within a model are sorted by ID, residues with a chain sorted by number, and atoms within a residue sorted by name before pairing.
Harper ________________________________ From: Roden Deng Luo via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Sent: Saturday, June 29, 2024 2:59 AM To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: [chimerax-users] RMSD does not make sense in a few cases
Hello ChimeraX Team,
In the attached screenshot, #1 and #3 are almost overlapping, but get an RMSD as 57. I occasionally encounter this. The session and log files are attached. In short, to reproduce, open 5nl2.cif and emd_3658.map; fitmap #1 in #2 search 1000; rmsd #1 to #3. This happened rarely, but in this case, it happened twice. It might be that I fit other structures first. But I indeed click "Clear List" and "close session" after each fitting. The second top hit's RMSD seems to make sense in the attached session (screenshot attached too).
Thanks, Roden
--
This message and its contents, including attachments are intended solely for the original recipient. If you are not the intended recipient or have received this message in error, please notify me immediately and delete this message from your computer system. Any unauthorized use or distribution is prohibited. Please consider the environment before printing this email.
-- This message and its contents, including attachments are intended solely for the original recipient. If you are not the intended recipient or have received this message in error, please notify me immediately and delete this message from your computer system. Any unauthorized use or distribution is prohibited. Please consider the environment before printing this email.
participants (3)
-
 Roden Deng Luo
Roden Deng Luo -
 Smith, Harper
Smith, Harper -
 Tom Goddard
Tom Goddard