OpenFold Tool Model Selection Question

Hello, I’ve recently started using the OpenFold tool in ChimeraX and I’ve found it really useful so far. I’m trying to model multimeric complexes and I’m wondering if the tool automatically detects multimeric inputs and uses OpenFold Multimer as the model? I noticed in some config files there is code that detects monomeric versus multimeric input, but I don’t see any indication of what model is used in any of my output files. Any input would be greatly appreciated. Thanks! Zach Eidman

Hi Zach, ChimeraX installs and runs predictions with OpenFold 3 preview 2 an implementation of AlphaFold 3. It does not use OpenFold 2 or OpenFold Multimer which are older programs based on AlphaFold 2. As far as I know there are no special settings for running multimer predictions in OpenFold 3 or AlphaFold 3. You specify all of your protein sequences and it will predict the complex. More info about ChimeraX OpenFold 3 capabilities can be found here https://www.rbvi.ucsf.edu/chimerax/data/openfold-feb2026/openfold.html Tom
On Apr 1, 2026, at 11:59 AM, Eidman, Zachary M via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I’ve recently started using the OpenFold tool in ChimeraX and I’ve found it really useful so far. I’m trying to model multimeric complexes and I’m wondering if the tool automatically detects multimeric inputs and uses OpenFold Multimer as the model? I noticed in some config files there is code that detects monomeric versus multimeric input, but I don’t see any indication of what model is used in any of my output files. Any input would be greatly appreciated.
Thanks! Zach Eidman _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/

In connection to the message below, in particular regarding the modeling of protein-ligand complexes, are there any advantages/disadvantages of OpenFold3 vs Boltz2? Thank you for your suggestions
On 1 Apr 2026, at 21:13, Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Zach,
ChimeraX installs and runs predictions with OpenFold 3 preview 2 an implementation of AlphaFold 3. It does not use OpenFold 2 or OpenFold Multimer which are older programs based on AlphaFold 2. As far as I know there are no special settings for running multimer predictions in OpenFold 3 or AlphaFold 3. You specify all of your protein sequences and it will predict the complex.
More info about ChimeraX OpenFold 3 capabilities can be found here
https://www.rbvi.ucsf.edu/chimerax/data/openfold-feb2026/openfold.html
Tom
On Apr 1, 2026, at 11:59 AM, Eidman, Zachary M via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I’ve recently started using the OpenFold tool in ChimeraX and I’ve found it really useful so far. I’m trying to model multimeric complexes and I’m wondering if the tool automatically detects multimeric inputs and uses OpenFold Multimer as the model? I noticed in some config files there is code that detects monomeric versus multimeric input, but I don’t see any indication of what model is used in any of my output files. Any input would be greatly appreciated.
Thanks! Zach Eidman _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
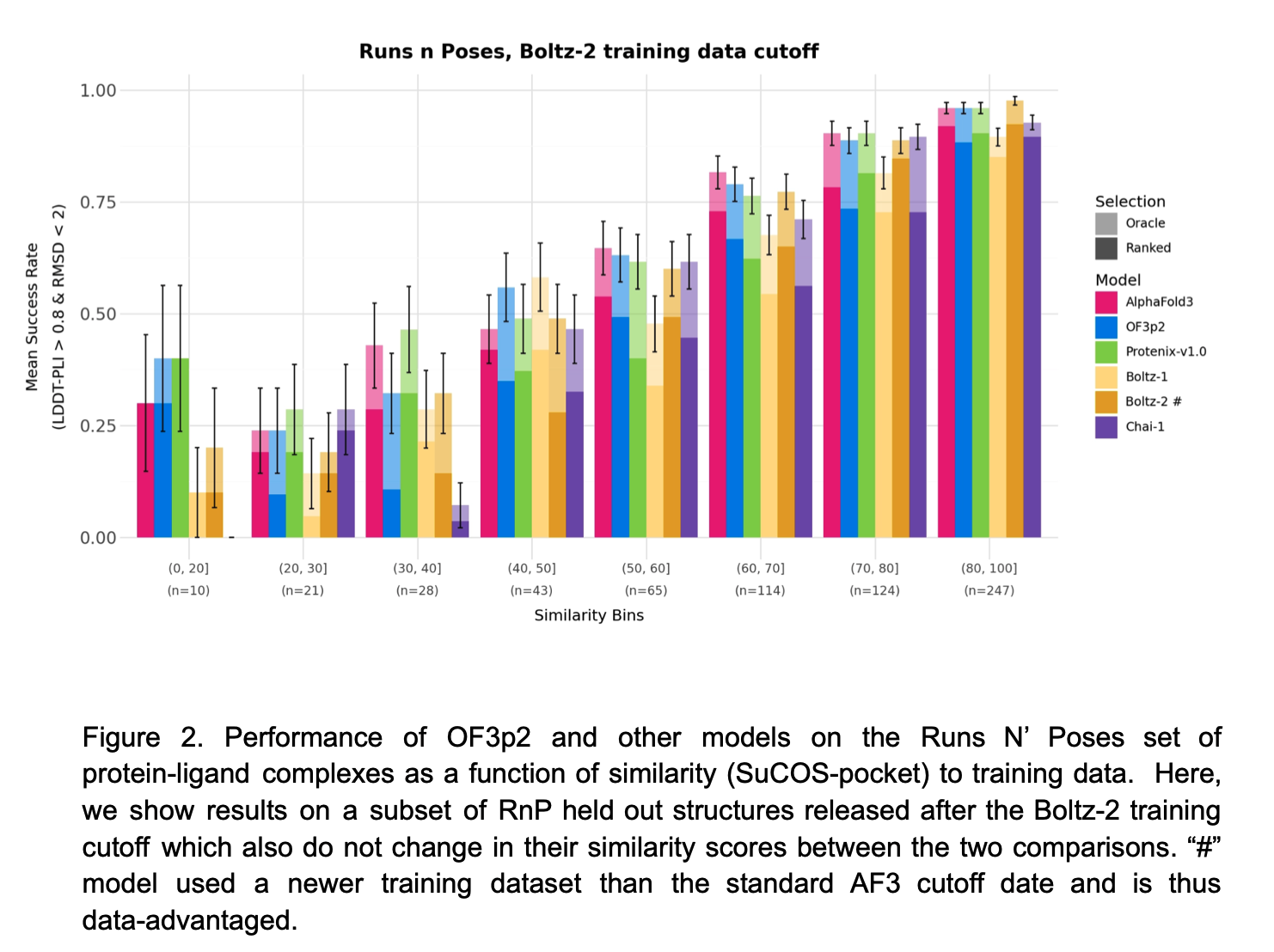
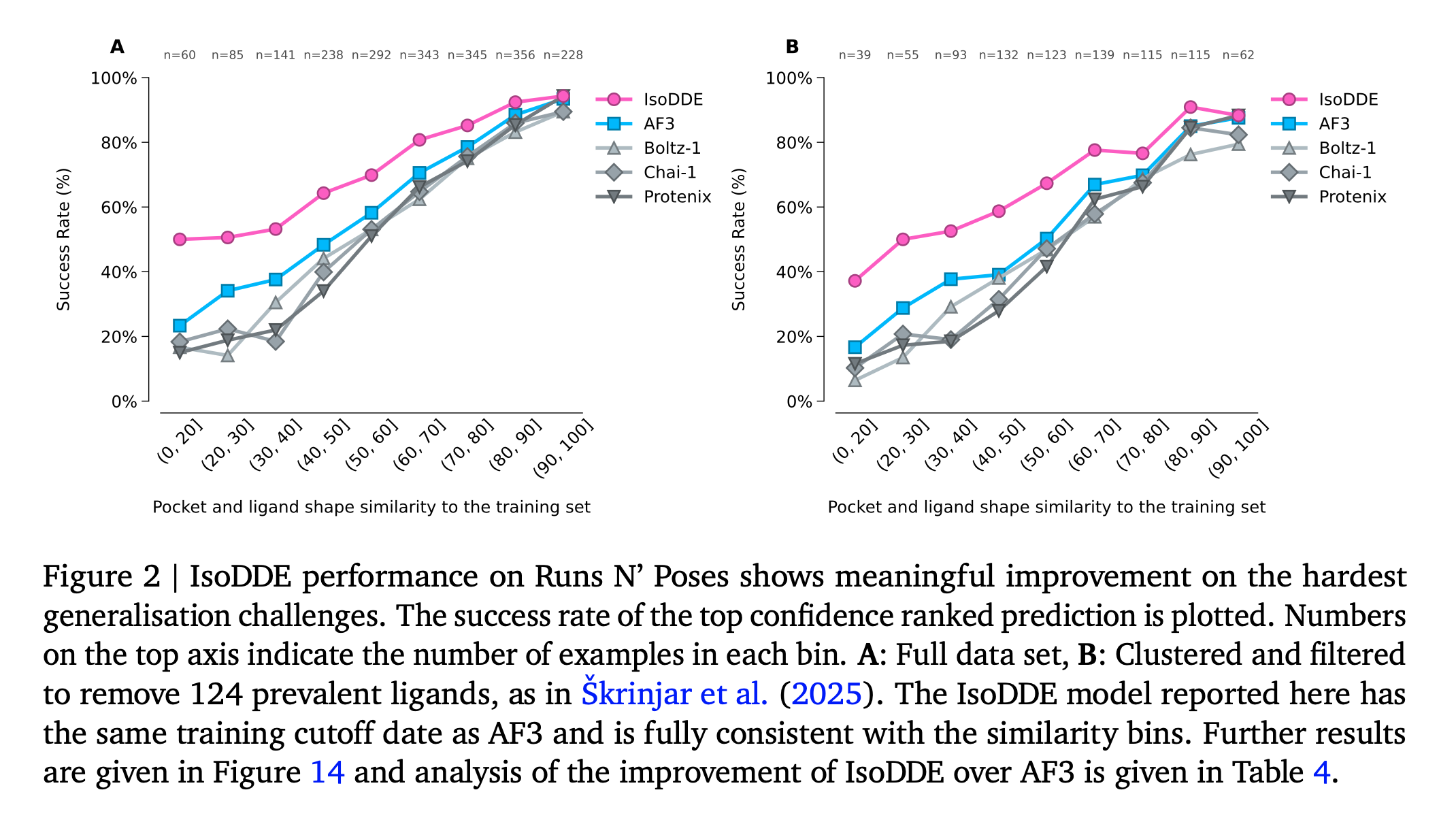
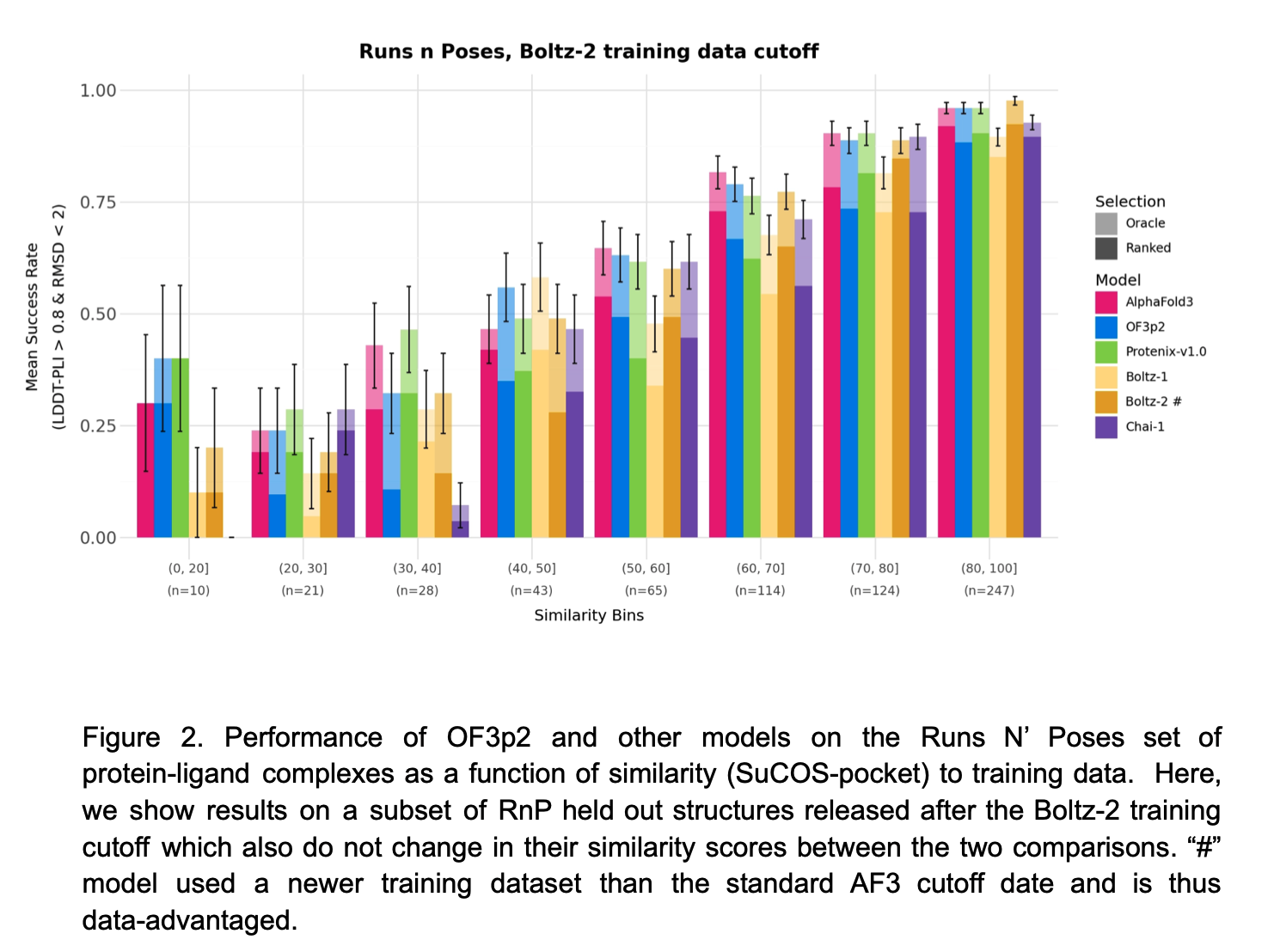
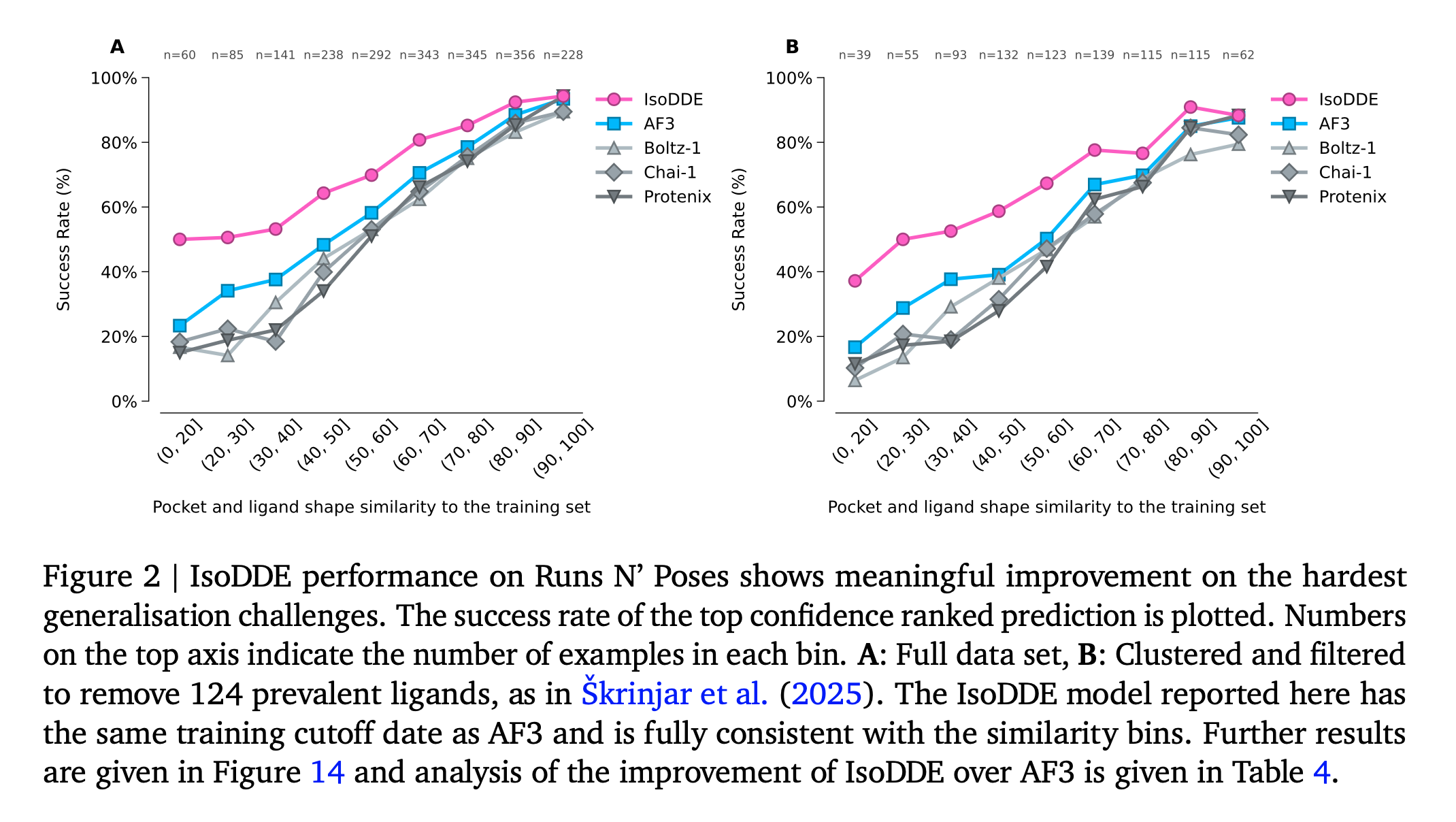
Hi George, Boltz 2 has the advantage that it can predict binding affinity. OpenFold 3 has an extension called AQAffinity that does the same prediction of binding affinity using the same method as Boltz 2 but I have not yet included it in ChimeraX. The binding affinity predictions are not very reliable. I am working mostly on OpenFold 3 in ChimeraX because the MIT development team that created Boltz started a company and has not made any Boltz improvements since September, and I think Boltz 2 will no longer be developed. I expect the structure predictions program to improve significantly in coming years and OpenFold 3 is actively developed so I think that is a better program to promote in ChimeraX. Here is a technical report on the current state of OpenFold 3 (March 2026) https://portal.openfold.omsf.io/reports/of3p2_technical_report.pdf Figure 2 (attached below) compares predictions of protein-ligand complexes made by OpenFold 3, Boltz 2, AlphaFold 3 and other programs. When the ligand is not similar to those found in training data the prediction quality is much worse. The figure suggests AlphaFold 3 is still a little better than the competition. Isomorphic Labs, the company created by Google to continue AlphaFold 3 work for drug development issued a white paper Februrary 2026 on their improved results with the Isomorphic Labs Drug Design Engine (IsoDDE aka "AlphaFold 4"). https://zenodo.org/records/18606681 They claim it has vastly better protein-ligand binding predictions than AlphaFold 3, shown in figure 2 of their white paper (attached below). But it is proprietary and you will probably never have access to that method unless you are a customer of Isomorphic Labs. Tom Figure 2 from Openfold 3 preview 2 white paper, March 2026  Figure 2 from IsoDDE white paper showing IsoDDE predicts protein-ligand interactions better especially when ligands and pockets are not similar to training data. 
On Apr 2, 2026, at 1:27 AM, George Tzotzos via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
In connection to the message below, in particular regarding the modeling of protein-ligand complexes, are there any advantages/disadvantages of OpenFold3 vs Boltz2?
Thank you for your suggestions
On 1 Apr 2026, at 21:13, Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Zach,
ChimeraX installs and runs predictions with OpenFold 3 preview 2 an implementation of AlphaFold 3. It does not use OpenFold 2 or OpenFold Multimer which are older programs based on AlphaFold 2. As far as I know there are no special settings for running multimer predictions in OpenFold 3 or AlphaFold 3. You specify all of your protein sequences and it will predict the complex.
More info about ChimeraX OpenFold 3 capabilities can be found here
https://www.rbvi.ucsf.edu/chimerax/data/openfold-feb2026/openfold.html
Tom
On Apr 1, 2026, at 11:59 AM, Eidman, Zachary M via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I’ve recently started using the OpenFold tool in ChimeraX and I’ve found it really useful so far. I’m trying to model multimeric complexes and I’m wondering if the tool automatically detects multimeric inputs and uses OpenFold Multimer as the model? I noticed in some config files there is code that detects monomeric versus multimeric input, but I don’t see any indication of what model is used in any of my output files. Any input would be greatly appreciated.
Thanks! Zach Eidman _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
{kind=link}
Thank you very much Tom for the very comprehensive and most useful answer George
On 2 Apr 2026, at 19:14, Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi George,
Boltz 2 has the advantage that it can predict binding affinity. OpenFold 3 has an extension called AQAffinity that does the same prediction of binding affinity using the same method as Boltz 2 but I have not yet included it in ChimeraX. The binding affinity predictions are not very reliable. I am working mostly on OpenFold 3 in ChimeraX because the MIT development team that created Boltz started a company and has not made any Boltz improvements since September, and I think Boltz 2 will no longer be developed. I expect the structure predictions program to improve significantly in coming years and OpenFold 3 is actively developed so I think that is a better program to promote in ChimeraX. Here is a technical report on the current state of OpenFold 3 (March 2026)
https://portal.openfold.omsf.io/reports/of3p2_technical_report.pdf
Figure 2 (attached below) compares predictions of protein-ligand complexes made by OpenFold 3, Boltz 2, AlphaFold 3 and other programs. When the ligand is not similar to those found in training data the prediction quality is much worse. The figure suggests AlphaFold 3 is still a little better than the competition.
Isomorphic Labs, the company created by Google to continue AlphaFold 3 work for drug development issued a white paper Februrary 2026 on their improved results with the Isomorphic Labs Drug Design Engine (IsoDDE aka "AlphaFold 4").
https://zenodo.org/records/18606681
They claim it has vastly better protein-ligand binding predictions than AlphaFold 3, shown in figure 2 of their white paper (attached below). But it is proprietary and you will probably never have access to that method unless you are a customer of Isomorphic Labs.
Tom
Figure 2 from Openfold 3 preview 2 white paper, March 2026 
Figure 2 from IsoDDE white paper showing IsoDDE predicts protein-ligand interactions better especially when ligands and pockets are not similar to training data. 
On Apr 2, 2026, at 1:27 AM, George Tzotzos via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
In connection to the message below, in particular regarding the modeling of protein-ligand complexes, are there any advantages/disadvantages of OpenFold3 vs Boltz2?
Thank you for your suggestions
On 1 Apr 2026, at 21:13, Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Zach,
ChimeraX installs and runs predictions with OpenFold 3 preview 2 an implementation of AlphaFold 3. It does not use OpenFold 2 or OpenFold Multimer which are older programs based on AlphaFold 2. As far as I know there are no special settings for running multimer predictions in OpenFold 3 or AlphaFold 3. You specify all of your protein sequences and it will predict the complex.
More info about ChimeraX OpenFold 3 capabilities can be found here
https://www.rbvi.ucsf.edu/chimerax/data/openfold-feb2026/openfold.html
Tom
On Apr 1, 2026, at 11:59 AM, Eidman, Zachary M via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I’ve recently started using the OpenFold tool in ChimeraX and I’ve found it really useful so far. I’m trying to model multimeric complexes and I’m wondering if the tool automatically detects multimeric inputs and uses OpenFold Multimer as the model? I noticed in some config files there is code that detects monomeric versus multimeric input, but I don’t see any indication of what model is used in any of my output files. Any input would be greatly appreciated.
Thanks! Zach Eidman _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
{kind=link}
participants (3)
-
 Eidman, Zachary M
Eidman, Zachary M -
 George Tzotzos
George Tzotzos -
 Tom Goddard
Tom Goddard