Coloring of trajectory using mpl command

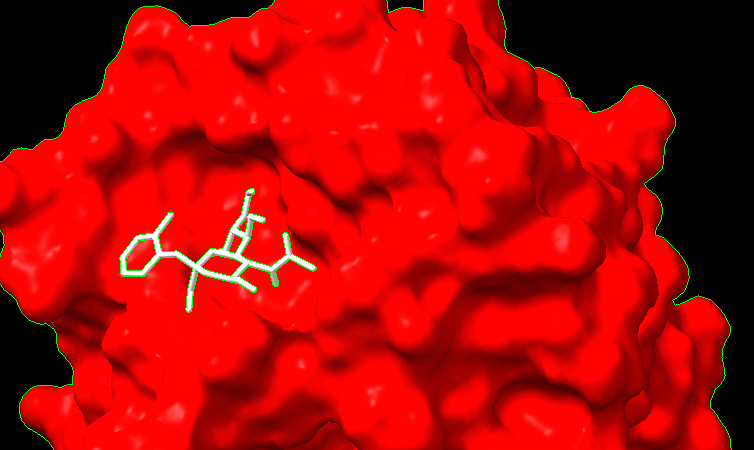
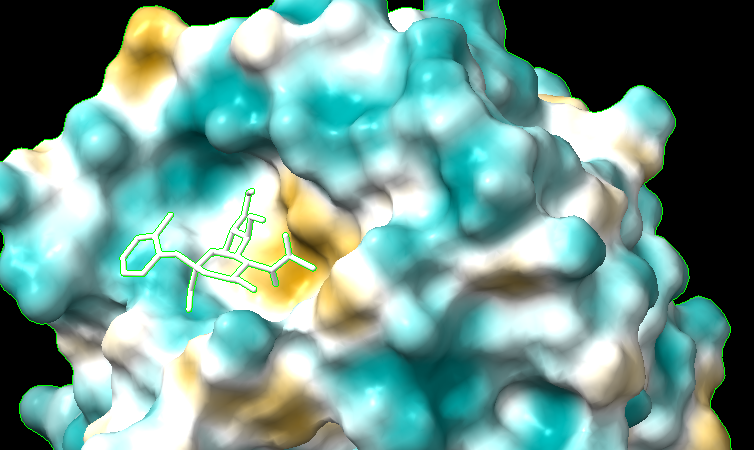
Hello everyone, I'm sorry for bothering all of you so often these past few days... but I ran into another issue. I opened a pdb and corresponding trajectory .nc file, and I ran "mlp #1 key true". Lipophilicity coloring showed up, but when I played the trajectory, the mpl colors went away and the default colors came back. I noticed the same happened for "coulombic" command. For now, I'm making due with a custom attribute file and "color byattribute name palette lipophilicity" from this tutorial https://www.cgl.ucsf.edu/chimerax/docs/user/formats/defattr.html#examples. It does stay throughout the trajectory but doesn't look as good, and the colors aren't blended. I have also tried doing "mlp map true" and "color sample #1 map #2" with #1 being the pdb with trajectory and #2 the resulting map from mlp. It looked really bad. The protein is now one color. "mlp map true" command [cid:41c39c60-61c8-47ff-b996-f063b013d319] [cid:d2f10dad-4f81-40e6-99cc-407b2d96a17f] [cid:ec390e08-8756-45ce-af0d-8e8a03992457] Alex
{kind=link}
{kind=link}
{kind=link}

Hi Alex, The coulombic and mlp commands work by: (1) calculating a spatial distribution of potential values (electrostatic or lipophilic) in 3D using atomic positions and atomic parameters (charges or lipophilicities) (2) determining the resulting potential at or near the position of each surface vertex and then applying surface coloring accordingly In a trajectory both the atomic positions and the positions of the surface vertices are changing, so you would need to run the calculation again for each frame of the trajectory, i.e. each time the set of atomic coordinates changes. More on that below. This problem of needing to run a new calculation when the coordinates change is equally true regardless of if you open the potential from step 1 above as a map model and apply coloring separately with "color sample," so there is no reason to do that, which just makes things more complicated. Your "color sample" command made the surface red because you did not specify the palette (what colors to use for what values) and it used the default, which is red-white-blue across the whole range of values. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/color.html#map> If you wanted the coloring to look like mlp coloring you would need to use with "color sample" the same palette settings that "mlp" uses: palette lipophilicity range -20,20 ...as mentioned in the mlp docs: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/mlp.html#palette-options> ...so it would be something like color sample #1 map #2 palette lipophilicity range -20,20 However, as explained above, you don't need to bother with creating a map model and doing coloring in a separate step. You just need to run "mlp" at each trajectory frame using "perframe" as in this example: open 1plx coordset true perframe "coordset #1 $1 ; surf close ; surf #1 update false ; mlp surf #1" range 1,80 For technical reasons I won't get into, unfortunately this command is more convoluted than it should be, requiring deleting and remaking the surface in addition to re-running mlp at each trajectory frame. Attribute coloring, on the other hand, is quite different than coulombic or mlp coloring. It doesn't use an equation to compute values at the surface vertices based on the coordinates of all the nearby atoms. Instead you simply assign a constant value per atom or per residue from a lookup table (such as the amino acid hydrophobicity scales in the example attribute files). Then the molecular surface patch for that atom or residue will be colored by that constant value from the lookup table. That is why you don't get blending. There is one constant value per residue, in the case of amino acid hydrophobicity, and abrupt boundaries between the surface patches of different residues. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/color.html#byattribute> <https://rbvi.ucsf.edu/chimerax/docs/user/attributes.html> I hope this helps and explains some of your observations. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 7, 2023, at 9:34 AM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello everyone,
I'm sorry for bothering all of you so often these past few days... but I ran into another issue. I opened a pdb and corresponding trajectory .nc file, and I ran "mlp #1 key true". Lipophilicity coloring showed up, but when I played the trajectory, the mpl colors went away and the default colors came back. I noticed the same happened for "coulombic" command. For now, I'm making due with a custom attribute file and "color byattribute name palette lipophilicity" from this tutorialhttps://www.cgl.ucsf.edu/chimerax/docs/user/formats/defattr.html#examples. It does stay throughout the trajectory but doesn't look as good, and the colors aren't blended.
I have also tried doing "mlp map true" and "color sample #1 map #2" with #1 being the pdb with trajectory and #2 the resulting map from mlp. It looked really bad. The protein is now one color. "mlp map true" command <image.png> <image.png> <image.png>
Alex _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
participants (2)
-
 Alexander Hung Lee
Alexander Hung Lee -
 Elaine Meng
Elaine Meng