Questions on Visualizing Chromatin Structure and Gene Expression in ChimeraX

Dear UCSF ChimeraX Team, I am a big fan of your program and I have been greatly impressed with its capabilities. I want to express my deep appreciation for the "soft" visualization option provided in ChimeraX - the aesthetics truly appeal to me. I've been utilizing ChimeraX to visualize my chromatin structure data. While I'm aware this might not be one of the intended uses, when I simulated the 3D structure of chromatin as a protein's cif file, the result looked fantastic. Now I've encountered a challenge and I am hopeful that you might be able to provide some guidance. I am looking to represent gene expression as the size of the spheres in my chromatin structure, all in one figure. I am aware that Chimera has a method using "vdwdefine", essentially using the size of the B-factor to represent the atomic radius. Could you guide me on how to achieve this in ChimeraX? Furthermore, if I want to include multiple columns similar to the B-factor, how can I add them to the cif? Using the "defattr" command feels a bit cumbersome. It would be very convenient if I could pack a lot of dimensional information (such as the gene density of this chromatin segment, CG content, chromatin opening degree, etc.) into a single cif file. Do you have any suggestions or strategies on how to achieve these somewhat unconventional functionalities? Your assistance would be greatly appreciated. Thank you for your attention and I look forward to hearing from you soon. Best Regards, Zhiyuan Liu
{kind=link}
{kind=link}

HI Zhiyuan, Those genome packaging images are cool looking. If your genome segments are represented by a single PDB atom and the B-factor value represents gene expression or some other quantity of interest, you can control the atom color using the ChimeraX Render by Attribute tool or "color byattribute" tool. Those cannot change the atom radius. The ChimeraX size command can set the atom radius, but it can't use a formula related to bfactor. So to set the size of an atom according to its bfactor you would need to use a little Python code. This code sets the radius of each atom to 1.0 plus 0.1 times the bfactor. from chimerax.atomic import Structure models = session.models.list(type = Structure) for m in models: for a in m.atoms: a.radius = 1.0 + 0.1 * a.bfactor If you put it in a file bsize.py and open that file in ChimeraX it will run it and set the atom sizes. You need to show the atoms in spheres style (not stick) to see this radius, command "style sphere". It would be nicer to write a bit more Python that makes this a command you can type to ChimeraX like "atomsize #1 attribute bfactor scale 0.1 offset 1.0". Then you could easily try different scale factors and also use different attributes. I'll make some example Python code for that later today and send a link. To add more numeric attributes for each atom you can use a ChimeraX attribute file with lines like :1 27.5 :2 10.8 ... Examples are in the ChimeraX documentation. While you could put custom attributes into an mmCIF file in the _atom_site table ChimeraX has no builtin capabilities to recognize custom attributes. It would be possible to write a little Python code that reads those attributes from mmCIF and assigns them as Python attributes of the atoms that you could then use for coloring or sizing. But you would also need custom code to write such an mmCIF file. The PDB started a repository for integrative hybrid models (derived from all different kinds of data) about 5 years ago. The repository is called PDBDev. I wrote some ChimeraX code to open those models. They intended to support genome packing models but I don't know if they every did. They have their own CIF dictionary for these models. You might look at that format rather than hacking the mmCIF format which is only intended for atomic resolution molecular structures. Tom
On Jul 22, 2023, at 8:55 AM, 刘致远 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear UCSF ChimeraX Team,
I am a big fan of your program and I have been greatly impressed with its capabilities. I want to express my deep appreciation for the "soft" visualization option provided in ChimeraX - the aesthetics truly appeal to me.
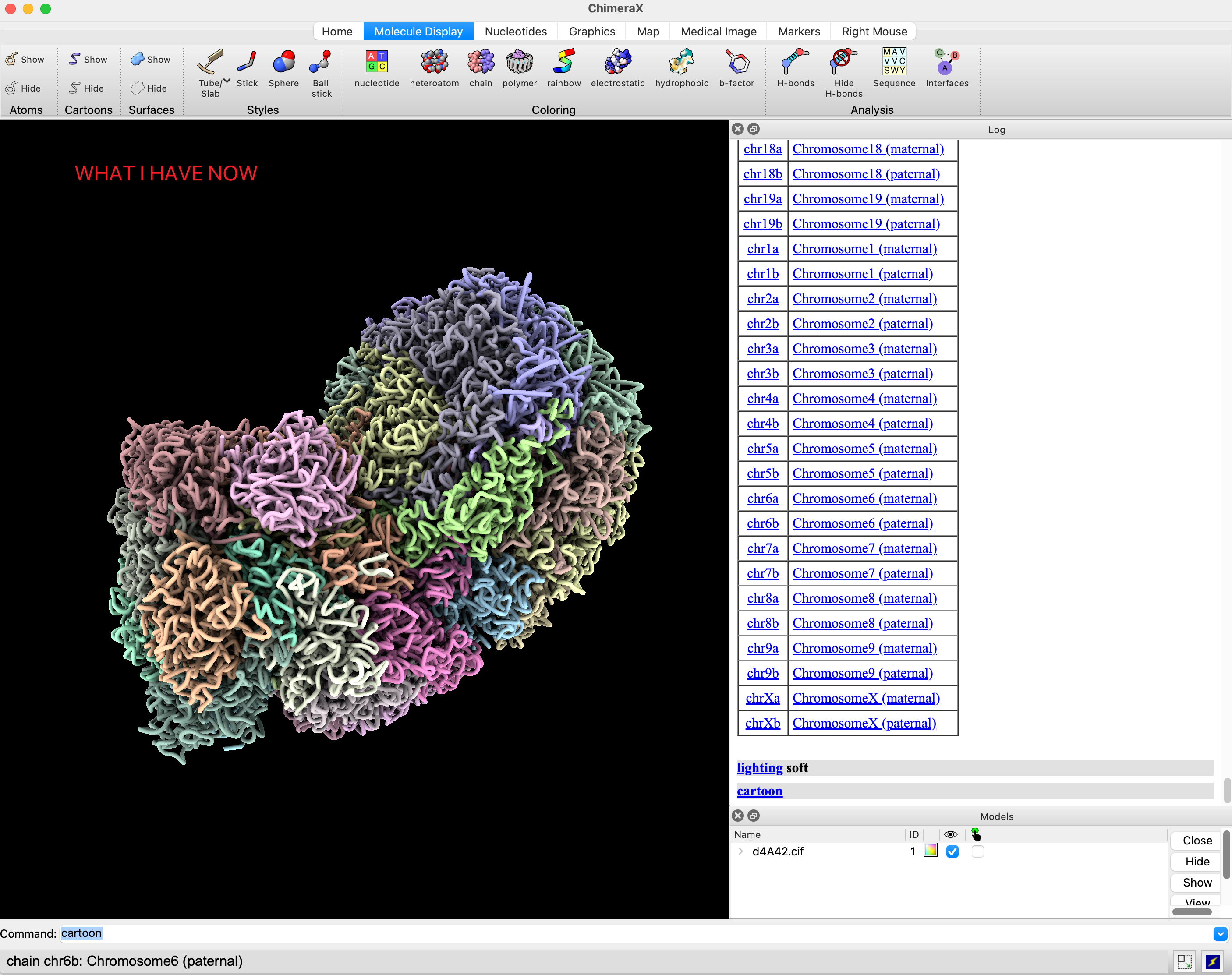
I've been utilizing ChimeraX to visualize my chromatin structure data. While I'm aware this might not be one of the intended uses, when I simulated the 3D structure of chromatin as a protein's cif file, the result looked fantastic.
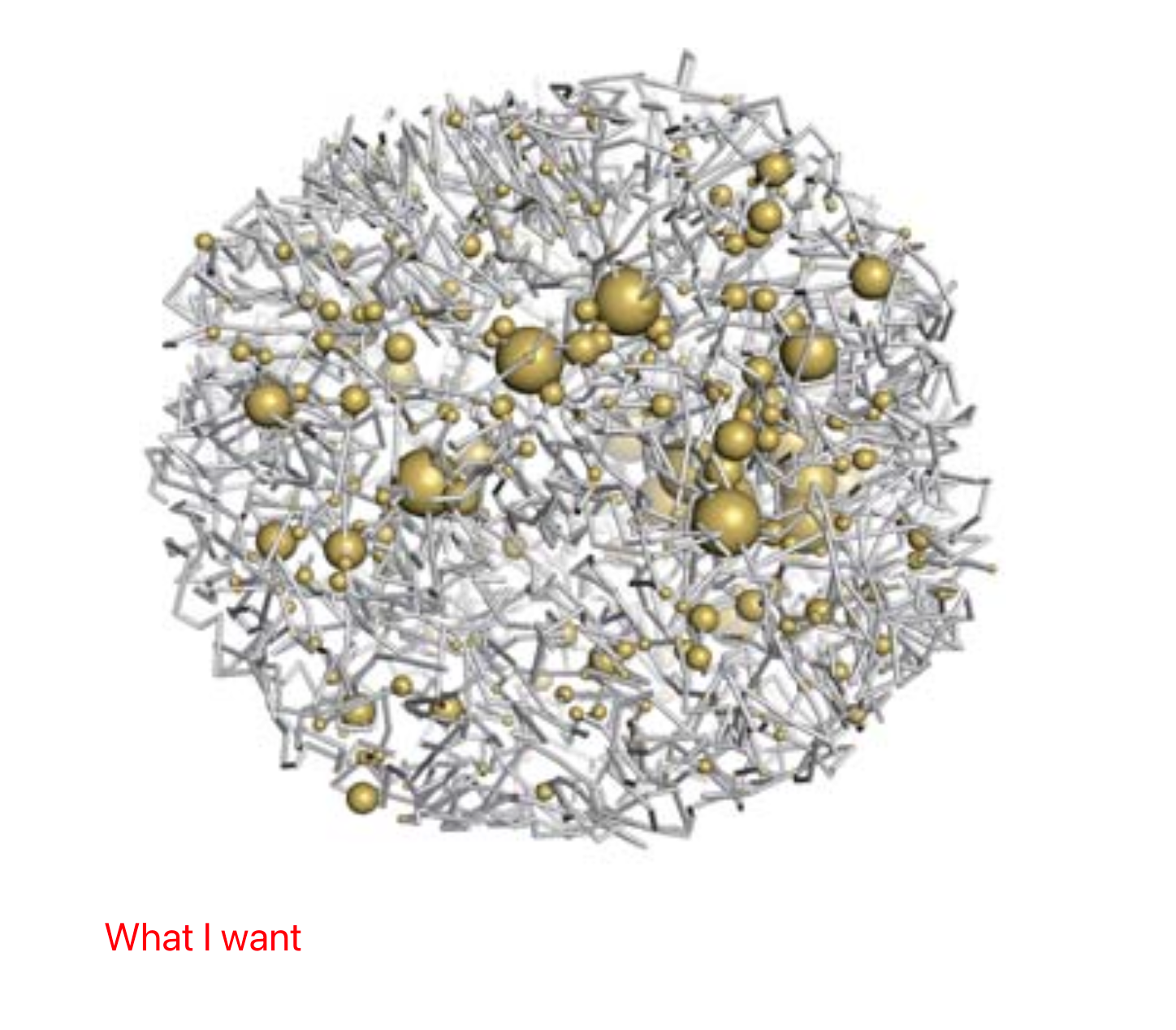
Now I've encountered a challenge and I am hopeful that you might be able to provide some guidance. I am looking to represent gene expression as the size of the spheres in my chromatin structure, all in one figure. I am aware that Chimera has a method using "vdwdefine", essentially using the size of the B-factor to represent the atomic radius. Could you guide me on how to achieve this in ChimeraX?
Furthermore, if I want to include multiple columns similar to the B-factor, how can I add them to the cif? Using the "defattr" command feels a bit cumbersome. It would be very convenient if I could pack a lot of dimensional information (such as the gene density of this chromatin segment, CG content, chromatin opening degree, etc.) into a single cif file.
Do you have any suggestions or strategies on how to achieve these somewhat unconventional functionalities? Your assistance would be greatly appreciated.
Thank you for your attention and I look forward to hearing from you soon.
Best Regards,
Zhiyuan Liu <image.png>
<image.png>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
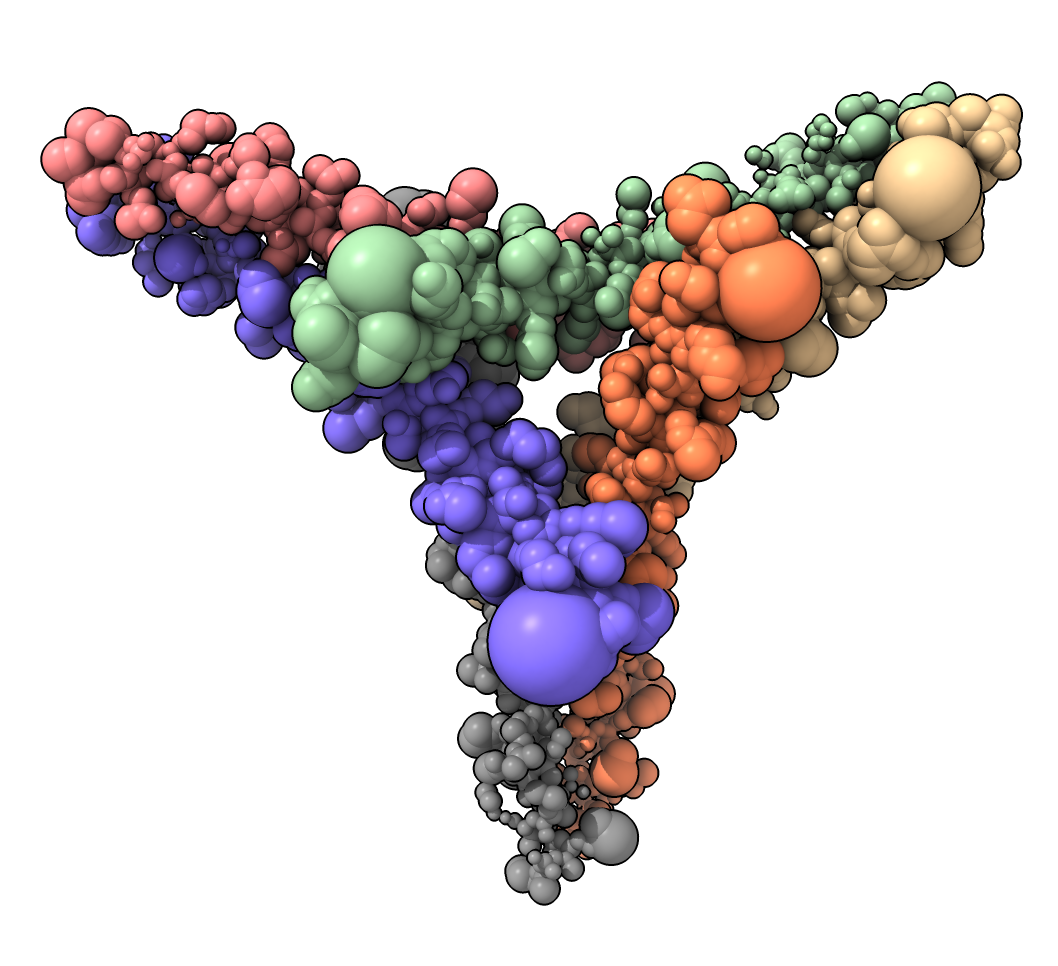
Hi Zhiyuan, Here's some Python code that creates a new ChimeraX command "atomsize" for setting the atom size proportional to an attribute value. https://rbvi.github.io/chimerax-recipes/atomsize/atomsize.html Tom "atomsize #1 scale 0.03" to PDB 7w91 
On Jul 24, 2023, at 12:22 PM, Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
HI Zhiyuan,
Those genome packaging images are cool looking. If your genome segments are represented by a single PDB atom and the B-factor value represents gene expression or some other quantity of interest, you can control the atom color using the ChimeraX Render by Attribute tool or "color byattribute" tool. Those cannot change the atom radius. The ChimeraX size command can set the atom radius, but it can't use a formula related to bfactor. So to set the size of an atom according to its bfactor you would need to use a little Python code. This code sets the radius of each atom to 1.0 plus 0.1 times the bfactor.
from chimerax.atomic import Structure models = session.models.list(type = Structure) for m in models: for a in m.atoms: a.radius = 1.0 + 0.1 * a.bfactor
If you put it in a file bsize.py and open that file in ChimeraX it will run it and set the atom sizes. You need to show the atoms in spheres style (not stick) to see this radius, command "style sphere". It would be nicer to write a bit more Python that makes this a command you can type to ChimeraX like "atomsize #1 attribute bfactor scale 0.1 offset 1.0". Then you could easily try different scale factors and also use different attributes. I'll make some example Python code for that later today and send a link.
To add more numeric attributes for each atom you can use a ChimeraX attribute file with lines like
:1 27.5 :2 10.8 ...
Examples are in the ChimeraX documentation.
While you could put custom attributes into an mmCIF file in the _atom_site table ChimeraX has no builtin capabilities to recognize custom attributes. It would be possible to write a little Python code that reads those attributes from mmCIF and assigns them as Python attributes of the atoms that you could then use for coloring or sizing. But you would also need custom code to write such an mmCIF file.
The PDB started a repository for integrative hybrid models (derived from all different kinds of data) about 5 years ago. The repository is called PDBDev. I wrote some ChimeraX code to open those models. They intended to support genome packing models but I don't know if they every did. They have their own CIF dictionary for these models. You might look at that format rather than hacking the mmCIF format which is only intended for atomic resolution molecular structures.
Tom
On Jul 22, 2023, at 8:55 AM, 刘致远 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear UCSF ChimeraX Team,
I am a big fan of your program and I have been greatly impressed with its capabilities. I want to express my deep appreciation for the "soft" visualization option provided in ChimeraX - the aesthetics truly appeal to me.
I've been utilizing ChimeraX to visualize my chromatin structure data. While I'm aware this might not be one of the intended uses, when I simulated the 3D structure of chromatin as a protein's cif file, the result looked fantastic.
Now I've encountered a challenge and I am hopeful that you might be able to provide some guidance. I am looking to represent gene expression as the size of the spheres in my chromatin structure, all in one figure. I am aware that Chimera has a method using "vdwdefine", essentially using the size of the B-factor to represent the atomic radius. Could you guide me on how to achieve this in ChimeraX?
Furthermore, if I want to include multiple columns similar to the B-factor, how can I add them to the cif? Using the "defattr" command feels a bit cumbersome. It would be very convenient if I could pack a lot of dimensional information (such as the gene density of this chromatin segment, CG content, chromatin opening degree, etc.) into a single cif file.
Do you have any suggestions or strategies on how to achieve these somewhat unconventional functionalities? Your assistance would be greatly appreciated.
Thank you for your attention and I look forward to hearing from you soon.
Best Regards,
Zhiyuan Liu <image.png>
<image.png>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
participants (2)
-
 Tom Goddard
Tom Goddard -
 刘致远
刘致远