Show side chain, main chain, and cartoon at the same time

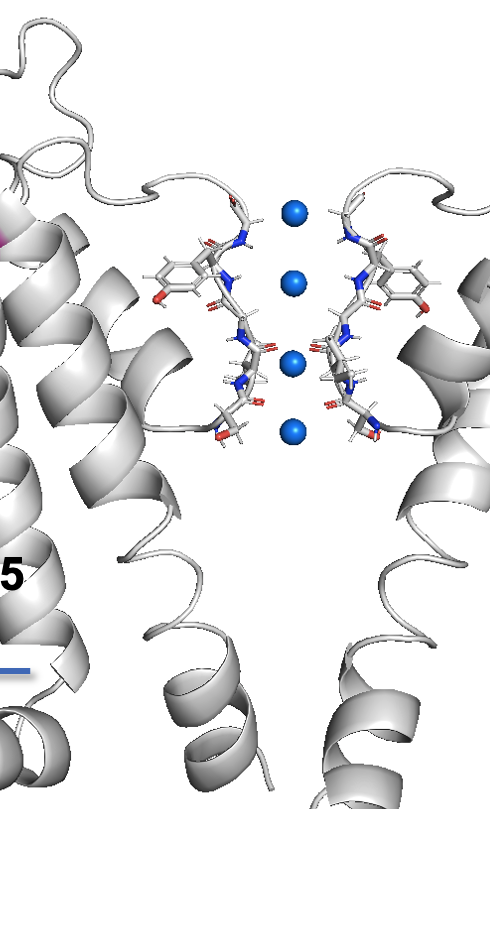
Dear colleagues I am new to ChimeraX. I am trying to figure out how to show side chain, main chain, and cartoon at the same time in a protein structure. Like the one below made in other software. Could you please help? [cid:image001.png@01D9D154.CE3D4440] Best, - - - - - - - - - - - - - - - - - Miao Zhang Ph.D. Associate Professor Chapman University School of Pharmacy 9401 Jeronimo Rd, Suite 297W Irvine, CA 92618 Phone: 714-516-5478 http://www.chapman.edu/our-faculty/miao-zhang
{kind=link}

Dear Miao Zhang, It is easy to show cartoons and sidechain atoms for any residues that you want (e.g. with "cartoon" and "show" commands or by clicking the toolbar icons that display cartoons and atoms, respectively). However, the tricky part is that if you want to also show mainchain, you have to use the option "suppress false" with "cartoon" command because normally cartoon will prevent showing mainchain. Here is an example to show sidechain + mainchain + cartoon in potassium channel structure 1bl8. Commands below with "..." to mean explanations that are not commands: open 1bl8 ... by default it already shows the atoms near the ions but you can hide all atoms first to start from a clean state hide ... select residues near ions (4.0 angstroms in this example) and then show their atoms ... at first only sidechain will be shown select ions :<4.0 show sel ... now use the cartoon option to also show mainchain at same time, and then clear the selection cartoon suppress false ~sel ... you could also just give chain ID and residue numbers instead of using a selection zone, for example residues 30-38 of chain D. The cartoon is already in the suppress-false state so you don't have to give that command again: show /D:30-38 ... Even though suppress false is turned on, you don't *have* to show mainchain. This setting just allows you to hide/show those atoms as you wish. So you can still hide the main chain atoms where you don't want to see them, for example: hide /D:30-38@n,c,o ... (you still need to keep the CA atoms shown or else the side chains will not be attached) See help pages for all the commands above in the User Guide for more details. <https://rbvi.ucsf.edu/chimerax/docs/user/index.html> You may also want to try some tutorials to become more familiar with ChimeraX: <https://www.rbvi.ucsf.edu/chimerax/tutorials.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 17, 2023, at 9:50 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear colleagues I am new to ChimeraX. I am trying to figure out how to show side chain, main chain, and cartoon at the same time in a protein structure. Like the one below made in other software. Could you please help? <image001.png> Best, - - - - - - - - - - - - - - - - - Miao Zhang Ph.D. Associate Professor Chapman University School of Pharmacy 9401 Jeronimo Rd, Suite 297W Irvine, CA 92618 Phone: 714-516-5478 http://www.chapman.edu/our-faculty/miao-zhang
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Thanks so much. That worked. Another question. When colored by elements, the color of carbons in side chains is different from the cartoons. Any chance I can change the color of carbons to the same color as the cartoons. [cid:image001.png@01D9D477.8B757DF0] From: Elaine Meng <meng@cgl.ucsf.edu> Date: Friday, August 18, 2023 at 9:19 AM To: Zhang, Miao <zhang@chapman.edu> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Show side chain, main chain, and cartoon at the same time External Message [External Message] Dear Miao Zhang, It is easy to show cartoons and sidechain atoms for any residues that you want (e.g. with "cartoon" and "show" commands or by clicking the toolbar icons that display cartoons and atoms, respectively). However, the tricky part is that if you want to also show mainchain, you have to use the option "suppress false" with "cartoon" command because normally cartoon will prevent showing mainchain. Here is an example to show sidechain + mainchain + cartoon in potassium channel structure 1bl8. Commands below with "..." to mean explanations that are not commands: open 1bl8 ... by default it already shows the atoms near the ions but you can hide all atoms first to start from a clean state hide ... select residues near ions (4.0 angstroms in this example) and then show their atoms ... at first only sidechain will be shown select ions :<4.0 show sel ... now use the cartoon option to also show mainchain at same time, and then clear the selection cartoon suppress false ~sel ... you could also just give chain ID and residue numbers instead of using a selection zone, for example residues 30-38 of chain D. The cartoon is already in the suppress-false state so you don't have to give that command again: show /D:30-38 ... Even though suppress false is turned on, you don't *have* to show mainchain. This setting just allows you to hide/show those atoms as you wish. So you can still hide the main chain atoms where you don't want to see them, for example: hide /D:30-38@n,c,o ... (you still need to keep the CA atoms shown or else the side chains will not be attached) See help pages for all the commands above in the User Guide for more details. <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Findex.html&data=05%7C01%7Czhang%40chapman.edu%7Caa41e63062f1441d9d1408dba006e0d1%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638279723631640988%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=dncoLgGU85m0WOevP4hUTcLTwQcdixHaWXQFeCvgVdI%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/index.html>> You may also want to try some tutorials to become more familiar with ChimeraX: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Fchimerax%2Ftutorials.html&data=05%7C01%7Czhang%40chapman.edu%7Caa41e63062f1441d9d1408dba006e0d1%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638279723631640988%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=dfczrkkWmRYhDTIUwXYzNH%2BcAacctTrElnyLyRfm4as%3D&reserved=0<https://www.rbvi.ucsf.edu/chimerax/tutorials.html>> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 17, 2023, at 9:50 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear colleagues I am new to ChimeraX. I am trying to figure out how to show side chain, main chain, and cartoon at the same time in a protein structure. Like the one below made in other software. Could you please help? <image001.png> Best, - - - - - - - - - - - - - - - - - Miao Zhang Ph.D. Associate Professor Chapman University School of Pharmacy 9401 Jeronimo Rd, Suite 297W Irvine, CA 92618 Phone: 714-516-5478 http://www.chapman.edu/our-faculty/miao-zhang
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Fmail.cgl.ucsf.edu%2Fmailman%2Farchives%2Flist%2Fchimerax-users%40cgl.ucsf.edu%2F&data=05%7C01%7Czhang%40chapman.edu%7Caa41e63062f1441d9d1408dba006e0d1%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638279723631640988%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=YIlzObRG02Z7rqNci8hpIoyX%2FPvOGmcdOaSd60opmfc%3D&reserved=0<https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/>
- NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.
{kind=link}
Dear Miao Zhang, If the cartoons are the model color, the easy way to keep the carbon atoms the same color is to avoid using "color byelement" and instead use "color byhet" which will color only the non-carbon atoms. But if it is too late for that and too late for undo, yes, you can color just the carbon atoms any color you want. I don't know how you colored the cartoons but you would need to know their color so you can use the same one. If the cartoons are "sky blue," then you could color carbon atoms the same way, for example, command: color C sky blue target a The "C" is the element symbol for carbon. If you don't know what color the cartoons are exactly (don't know name of color) you can try selecting part of the cartoon with Ctrl-click and then using the Selection Inspector tool (click green magnifing glass icon on top bar). In Selection Inspector, change from Inspect: Atoms to Inspect: Residues, which will then include "cartoon color" as a square. Click the square to open the system color editor showing that color. Usually the system color editor will include some way of seeing the hex code, in this case #87CEEB, which could also be used in a command: color C #87CEEB target a You can color cartoons and atoms at the same time with "target ac": color C orange red target ac I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 21, 2023, at 9:37 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks so much. That worked. Another question. When colored by elements, the color of carbons in side chains is different from the cartoons. Any chance I can change the color of carbons to the same color as the cartoons. <image001.png>
How can I change the size of spheres? Two of them look too close, when shown with spheres. [cid:image001.png@01D9F25E.99BA47B0] From: Elaine Meng <meng@cgl.ucsf.edu> Date: Tuesday, August 22, 2023 at 7:59 AM To: Zhang, Miao <zhang@chapman.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Coloring carbons to match cartoon External Message [External Message] Dear Miao Zhang, If the cartoons are the model color, the easy way to keep the carbon atoms the same color is to avoid using "color byelement" and instead use "color byhet" which will color only the non-carbon atoms. But if it is too late for that and too late for undo, yes, you can color just the carbon atoms any color you want. I don't know how you colored the cartoons but you would need to know their color so you can use the same one. If the cartoons are "sky blue," then you could color carbon atoms the same way, for example, command: color C sky blue target a The "C" is the element symbol for carbon. If you don't know what color the cartoons are exactly (don't know name of color) you can try selecting part of the cartoon with Ctrl-click and then using the Selection Inspector tool (click green magnifing glass icon on top bar). In Selection Inspector, change from Inspect: Atoms to Inspect: Residues, which will then include "cartoon color" as a square. Click the square to open the system color editor showing that color. Usually the system color editor will include some way of seeing the hex code, in this case #87CEEB, which could also be used in a command: color C #87CEEB target a You can color cartoons and atoms at the same time with "target ac": color C orange red target ac I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 21, 2023, at 9:37 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks so much. That worked. Another question. When colored by elements, the color of carbons in side chains is different from the cartoons. Any chance I can change the color of carbons to the same color as the cartoons. <image001.png>
- NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.
{kind=link}
How do we show the disulfide bond in ChimeraX? We refined two structures from the same cryoEM dataset. One structure shows this specific disulfide bond directly in ChimeraX. The other does not show the disulfide bond. How can we force it to show? [cid:image002.png@01D9F29A.0E940F90] From: Zhang, Miao <zhang@chapman.edu> Date: Thursday, September 28, 2023 at 10:53 PM To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: Coloring carbons to match cartoon How can I change the size of spheres? Two of them look too close, when shown with spheres. [cid:image001.png@01D9F25E.99BA47B0] From: Elaine Meng <meng@cgl.ucsf.edu> Date: Tuesday, August 22, 2023 at 7:59 AM To: Zhang, Miao <zhang@chapman.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Coloring carbons to match cartoon External Message [External Message] Dear Miao Zhang, If the cartoons are the model color, the easy way to keep the carbon atoms the same color is to avoid using "color byelement" and instead use "color byhet" which will color only the non-carbon atoms. But if it is too late for that and too late for undo, yes, you can color just the carbon atoms any color you want. I don't know how you colored the cartoons but you would need to know their color so you can use the same one. If the cartoons are "sky blue," then you could color carbon atoms the same way, for example, command: color C sky blue target a The "C" is the element symbol for carbon. If you don't know what color the cartoons are exactly (don't know name of color) you can try selecting part of the cartoon with Ctrl-click and then using the Selection Inspector tool (click green magnifing glass icon on top bar). In Selection Inspector, change from Inspect: Atoms to Inspect: Residues, which will then include "cartoon color" as a square. Click the square to open the system color editor showing that color. Usually the system color editor will include some way of seeing the hex code, in this case #87CEEB, which could also be used in a command: color C #87CEEB target a You can color cartoons and atoms at the same time with "target ac": color C orange red target ac I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 21, 2023, at 9:37 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks so much. That worked. Another question. When colored by elements, the color of carbons in side chains is different from the cartoons. Any chance I can change the color of carbons to the same color as the cartoons. <image001.png>
- NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.
{kind=link}
{kind=link}
Dear Miao Zhang, (1) showing disulfide bond. ChimeraX shows the bond if it is described in your input data, but doesn't automatically add more bonds. So the data file that shows the bond must already have a LINK or CONECT for that bond, whereas the other file that does not have the bond information. However, you can add the bond yourself, for example, using the command: bond :cys@sg reasonable true ... if the distance is judged reasonable for the bond. This solution was mentioned on the chimera-users list if you want to see a specific example: <https://mail.cgl.ucsf.edu/mailman/archives/list/chimera-users@cgl.ucsf.edu/t...> (2) you can change the VDW radius of atom(s) in several ways, such as with the "size" command or using the Selection Inspector. Example commands using potassium channel: open 1bl8 sel K ... if you open the Selection Inspector (menu Actions... Inspect, or click green magnifying glass icon) you can see the current radius is 1.370. You can change the radius by changing the number in the Selection Inspector, or using the "size" command, for example: size sel atomRadius 1.2 <https://rbvi.ucsf.edu/chimerax/docs/user/tools/inspector.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/size.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 29, 2023, at 5:59 AM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
How do we show the disulfide bond in ChimeraX? We refined two structures from the same cryoEM dataset. One structure shows this specific disulfide bond directly in ChimeraX. The other does not show the disulfide bond. How can we force it to show?
<image002.png> From: Zhang, Miao <zhang@chapman.edu> Date: Thursday, September 28, 2023 at 10:53 PM To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: Coloring carbons to match cartoon
How can I change the size of spheres? Two of them look too close, when shown with spheres. <image001.png>
Dear Sir/Madam When I made a figure with part of the chain shown as sticks, and part of it shown as cartoons, there is a gap between the cartoon and sticks. Is there a way to fix it? [cid:image001.png@01DA0757.7436F980] Best, Miao
{kind=link}
Dear Miao, Details depend on whether the cartoon part is on the N-term side (lower residue numbers) or C-term side (higher residue numbers). If both, then you can combine the following answers. If cartoon is on the N-term side, for example, residues 1-9 of 1gcn, you could show the C atom of the last cartoon residue (so that there will be bond to N of the first atoms residue): open 1gcn hide cartoon cartoon :1-9 suppress false show :10-29 atoms show :9@c -- or maybe more atoms from residue 9 (if that looks better), for example -- show :9@c,ca The "suppress false" is important because it allows showing backbone atoms for the same residues that are also shown as cartoon. The full option name is "suppressBackboneDisplay" but you can use the shorter version. See the help if you want more explanation: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/cartoon.html> If cartoon is on the C-term side, for example, residues 10-29 of 1gcn, you could show the N atom of the first cartoon residue (so that there will be a bond to C of the last atoms residue): open 1gcn hide cartoon cartoon :10-29 suppress false show :1-9 atoms show :10@n -- or maybe more atoms, as above -- show :10@n,ca The reason to also show CA atoms from the cartoon residues is that the cartoon position will be close to CA but it might not be close to N or C. If the N or C is not close to the ribbon, you may see a small cone that connects it to the ribbon, which we call a "tether" .... and you can change tether color, shape, etc. or make it invisible (opacity 0 which means completely transparent) with the command "cartoon tether": <https://rbvi.ucsf.edu/chimerax/docs/user/commands/cartoon.html#tether> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 25, 2023, at 3:25 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Sir/Madam
When I made a figure with part of the chain shown as sticks, and part of it shown as cartoons, there is a gap between the cartoon and sticks. Is there a way to fix it?
<image001.png>
Best,
Miao
One minor correction... actually, you can't change the tether color -- it will be the same as the atom's color. Elaine
On Oct 25, 2023, at 3:58 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
The reason to also show CA atoms from the cartoon residues is that the cartoon position will be close to CA but it might not be close to N or C. If the N or C is not close to the ribbon, you may see a small cone that connects it to the ribbon, which we call a "tether" .... and you can change tether color, shape, etc. or make it invisible (opacity 0 which means completely transparent) with the command "cartoon tether": <https://rbvi.ucsf.edu/chimerax/docs/user/commands/cartoon.html#tether>
How do I change the size of label “5.6 angstrom” of distance? Miao From: Elaine Meng <meng@cgl.ucsf.edu> Date: Wednesday, October 25, 2023 at 4:03 PM To: Zhang, Miao <zhang@chapman.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Gap between cartoon and stick External Message [External Message] One minor correction... actually, you can't change the tether color -- it will be the same as the atom's color. Elaine
On Oct 25, 2023, at 3:58 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
The reason to also show CA atoms from the cartoon residues is that the cartoon position will be close to CA but it might not be close to N or C. If the N or C is not close to the ribbon, you may see a small cone that connects it to the ribbon, which we call a "tether" .... and you can change tether color, shape, etc. or make it invisible (opacity 0 which means completely transparent) with the command "cartoon tether": <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Fcartoon.html%23tether&data=05%7C01%7Czhang%40chapman.edu%7C1e482e6e50f84bd27d0708dbd5ae9f87%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638338718277512333%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=S7m0m4FTzXKC4WmBA7YcqgyWxzGfdmcxWpPZF2dXNgE%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/cartoon.html#tether>>
- NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.
Hi Miao, If you just want to make current labels bigger, either (A) menu: Actions... Label.. Set Label Height - or - (B) the "label" command "height" option, e.g. label height 2 (or whatever angstrom height you want). For details, see: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html> Or if you want to set a preference for labels created in the future, see "label defaultHeight" <https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html#defaultHeight> ...or, you can do the same thing with the Preferences GUI, the Labels section: <https://rbvi.ucsf.edu/chimerax/docs/user/preferences.html#labels> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 27, 2023, at 12:41 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
How do I change the size of label “5.6 angstrom” of distance? Miao
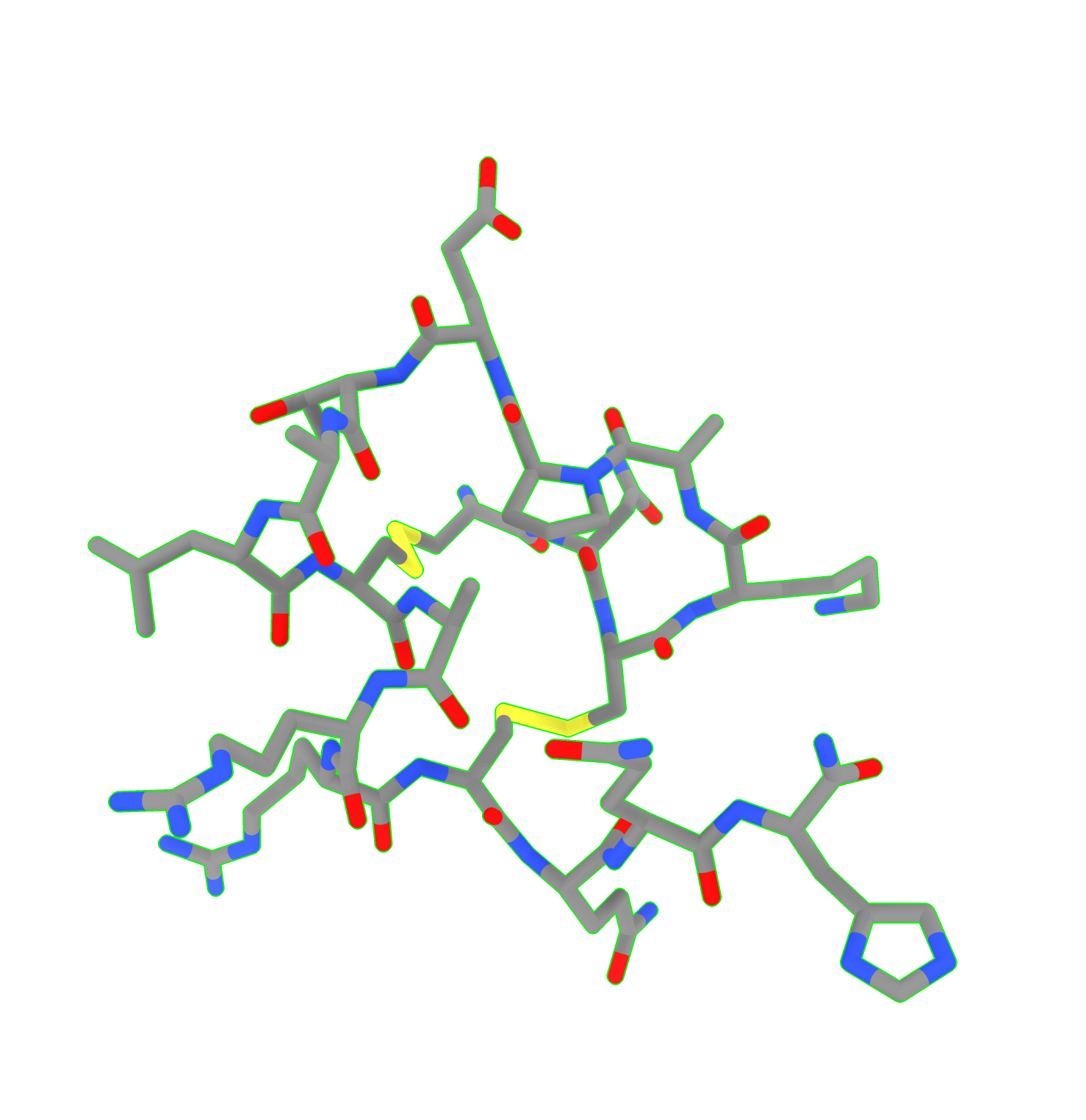
Dear Sir/Madam I have a small peptide that I want to show as cartoon. But chemira does not recognize it as a protein. I cannot show it as cartoon. I can only show it as surface, sphere, or sticks. How can I show cartoon? Many thanks Miao [cid:image001.png@01DA21EA.E87FBB80] From: Elaine Meng <meng@cgl.ucsf.edu> Date: Friday, October 27, 2023 at 1:08 PM To: Zhang, Miao <zhang@chapman.edu> Cc: Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Subject: change label size External Message [External Message] Hi Miao, If you just want to make current labels bigger, either (A) menu: Actions... Label.. Set Label Height - or - (B) the "label" command "height" option, e.g. label height 2 (or whatever angstrom height you want). For details, see: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Flabel.html&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=QEckL4Ww%2FgF0156cqf7N%2FOJ3lWa3VXY7c%2FTBt%2FMkD%2BI%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html>> Or if you want to set a preference for labels created in the future, see "label defaultHeight" <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Flabel.html%23defaultHeight&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=OMC1nNqy9ywqXAdyv1Kkpl6t%2BDTNzlPP1pnNI1JlnO4%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html#defaultHeight>> ...or, you can do the same thing with the Preferences GUI, the Labels section: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fpreferences.html%23labels&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=ey2EniLYdw9oSelNDM0%2Bmcf7jxJWJoe0aZcvkfs63ws%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/preferences.html#labels>> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 27, 2023, at 12:41 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
How do I change the size of label “5.6 angstrom” of distance? Miao
- NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.
{kind=link}
Hi Miao, I can't figure out the issue unless you also provide the example PDB file... a picture is not enough information. If OK to share this data with others, you can send it to chimerax-users, or alternatively just to me. Without the file, I can only guess that maybe the atom and residue names are not the usual atom and residue names for peptides/proteins. Thanks, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 28, 2023, at 11:06 AM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Sir/Madam I have a small peptide that I want to show as cartoon. But chemira does not recognize it as a protein. I cannot show it as cartoon. I can only show it as surface, sphere, or sticks. How can I show cartoon? Many thanks Miao <image001.png>
Summary of conclusions from seeing the data file: (1) In Miao's PDB file, the atoms had the correct names for a peptide, but the entire circular peptide was a single HET residue named UNK, instead of a series of standard amino acid residues with separate residue names and numbers. Thus ChimeraX could not recognize it as a peptide. (2) However, even for cyclic peptides that are in the correct format and are recognized as peptides (for example, 6ugc chain A), ChimeraX has a bug where it does not draw a closed loop for the cartoon. I think we have a ticket for this bug, although I failed to find it when I searched just now. Chimera does not have this bug, and will draw a closed loop ribbon for 6ugc chain A. Sorry about the difficulties, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 28, 2023, at 11:19 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Miao, I can't figure out the issue unless you also provide the example PDB file... a picture is not enough information. If OK to share this data with others, you can send it to chimerax-users, or alternatively just to me.
Without the file, I can only guess that maybe the atom and residue names are not the usual atom and residue names for peptides/proteins.
Thanks, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 28, 2023, at 11:06 AM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Sir/Madam I have a small peptide that I want to show as cartoon. But chemira does not recognize it as a protein. I cannot show it as cartoon. I can only show it as surface, sphere, or sticks. How can I show cartoon? Many thanks Miao <image001.png>
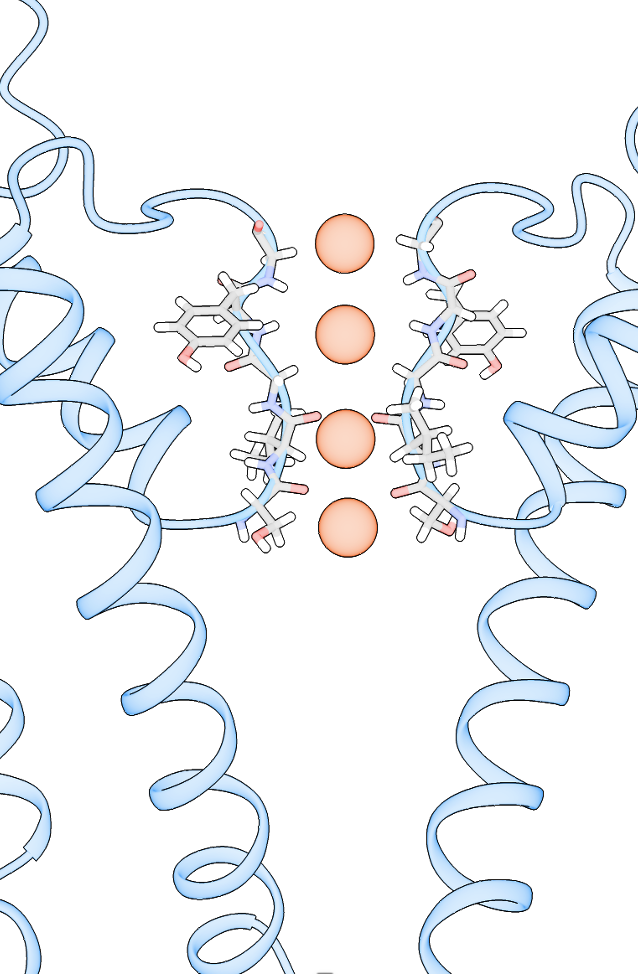
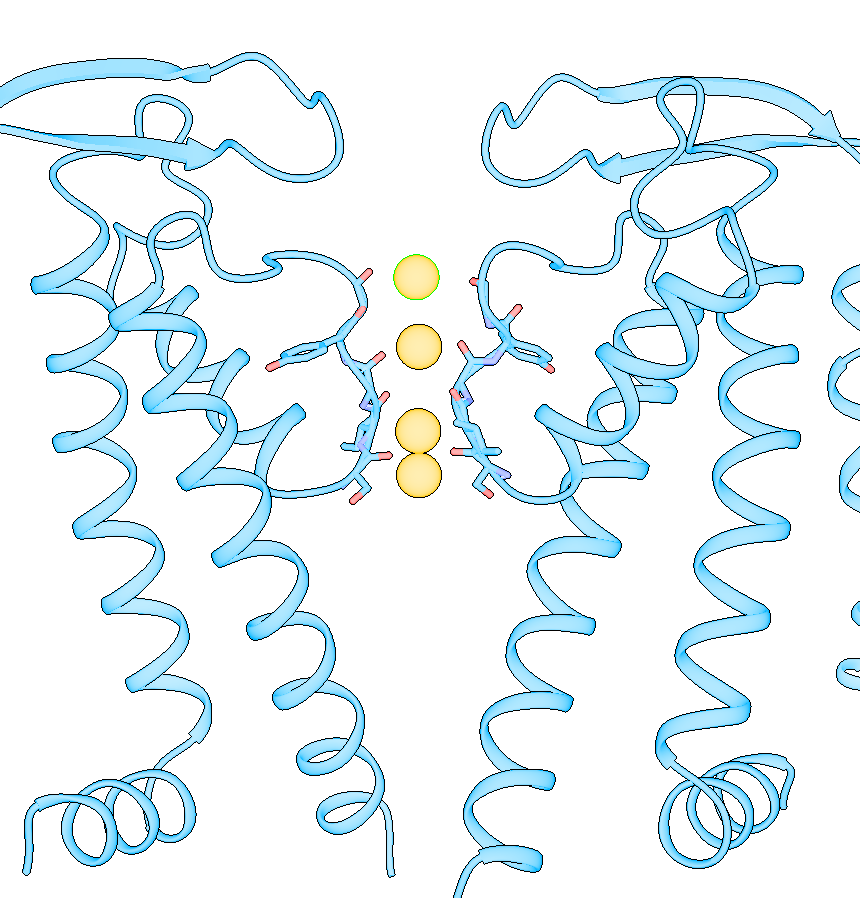
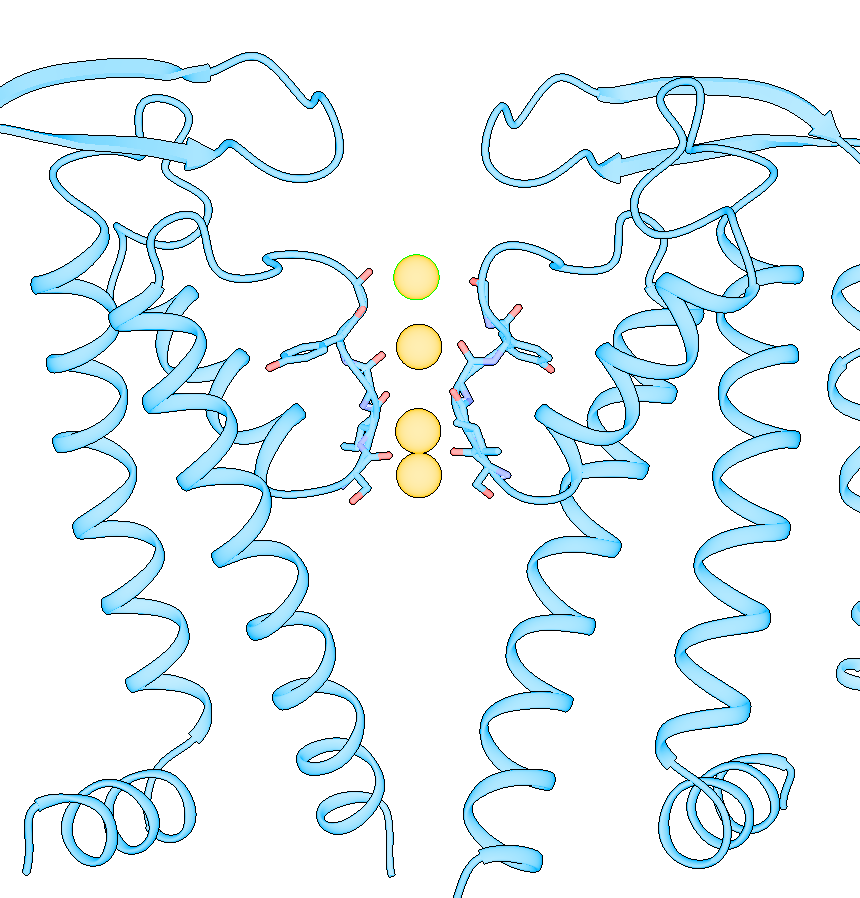
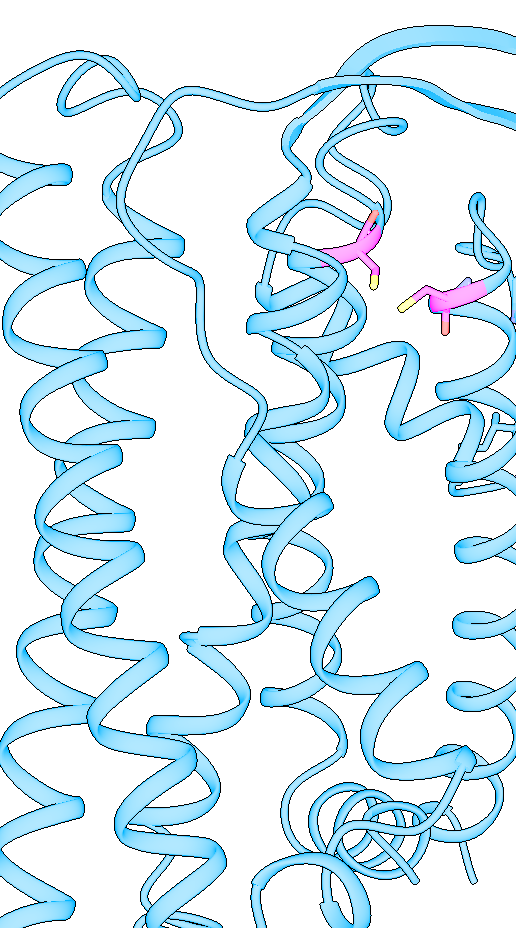
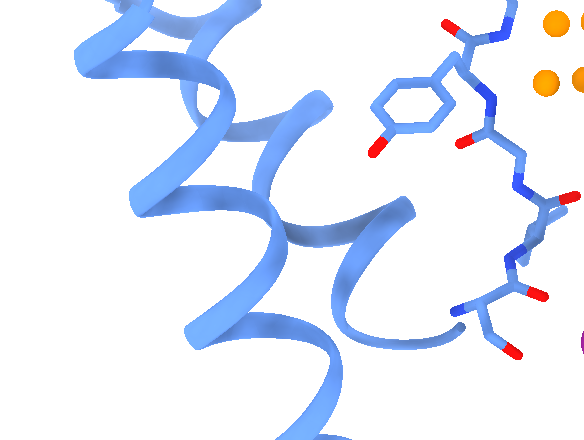
Dear Elaine We made figures using ChimeraX of the same protein. One structure with small molecule bound, one without. The cartoon style of the same region is shown differently in these two structures. How can we make them more consistent with each other? The cartoon style width is the same. [A close-up of a protein Description automatically generated] [A close-up of several spirals Description automatically generated] From: Elaine Meng <meng@cgl.ucsf.edu> Date: Friday, October 27, 2023 at 1:08 PM To: Zhang, Miao <zhang@chapman.edu> Cc: Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Subject: change label size External Message [External Message] Hi Miao, If you just want to make current labels bigger, either (A) menu: Actions... Label.. Set Label Height - or - (B) the "label" command "height" option, e.g. label height 2 (or whatever angstrom height you want). For details, see: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Flabel.html&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=QEckL4Ww%2FgF0156cqf7N%2FOJ3lWa3VXY7c%2FTBt%2FMkD%2BI%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html>> Or if you want to set a preference for labels created in the future, see "label defaultHeight" <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Flabel.html%23defaultHeight&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=OMC1nNqy9ywqXAdyv1Kkpl6t%2BDTNzlPP1pnNI1JlnO4%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html#defaultHeight>> ...or, you can do the same thing with the Preferences GUI, the Labels section: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fpreferences.html%23labels&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=ey2EniLYdw9oSelNDM0%2Bmcf7jxJWJoe0aZcvkfs63ws%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/preferences.html#labels>> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 27, 2023, at 12:41 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
How do I change the size of label “5.6 angstrom” of distance? Miao
- NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.
{kind=link}
{kind=link}
Hello, Most probably the issue is that some parts are not assigned as helix, so they are shown in the "coil" (non-helix, non-strand) ribbon style. In that case, it is because the backbone conformations of the two structures are slightly different and the H-bonding pattern is less ideal in some part, causing identification of that part as coil instead of helix. However, you can manually assign the residues as helix regardless of their conformation, if you really want to do that. In the assignment command, you would need to specify those residues by their residue number (and probably also chain ID and model number, if there are multiple chains and/or multiple models), for example: setattr #2/B:10-20 residues is_helix true see "setattr" help <https://rbvi.ucsf.edu/chimerax/docs/user/commands/setattr.html> You may also need to assign the attribute "ss_id" if you are still seeing short coil segments in the middle of where you want helix, as explained in this previous post: <https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/...> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 16, 2024, at 10:50 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Elaine We made figures using ChimeraX of the same protein. One structure with small molecule bound, one without. The cartoon style of the same region is shown differently in these two structures. How can we make them more consistent with each other? The cartoon style width is the same. <image001.png>
<image002.png>
Dear ChimeraX Help We made figures using ChimeraX of the same protein. One structure with small molecule bound, the other one without. The cartoon style of the same region is shown differently in these two structures. How can we make them more consistent with each other? The cartoon style width is the same. [A close-up of a protein Description automatically generated] [A close-up of several spirals Description automatically generated] From: Elaine Meng <meng@cgl.ucsf.edu> Date: Friday, October 27, 2023 at 1:08 PM To: Zhang, Miao <zhang@chapman.edu> Cc: Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Subject: change label size External Message [External Message] Hi Miao, If you just want to make current labels bigger, either (A) menu: Actions... Label.. Set Label Height - or - (B) the "label" command "height" option, e.g. label height 2 (or whatever angstrom height you want). For details, see: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Flabel.html&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=QEckL4Ww%2FgF0156cqf7N%2FOJ3lWa3VXY7c%2FTBt%2FMkD%2BI%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html>> Or if you want to set a preference for labels created in the future, see "label defaultHeight" <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Flabel.html%23defaultHeight&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=OMC1nNqy9ywqXAdyv1Kkpl6t%2BDTNzlPP1pnNI1JlnO4%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html#defaultHeight>> ...or, you can do the same thing with the Preferences GUI, the Labels section: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fpreferences.html%23labels&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=ey2EniLYdw9oSelNDM0%2Bmcf7jxJWJoe0aZcvkfs63ws%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/preferences.html#labels>> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 27, 2023, at 12:41 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
How do I change the size of label “5.6 angstrom” of distance? Miao
- NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.
{kind=link}
{kind=link}
Dear Miao Zhang, Please see previous answer to your same question from last week: <https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/...> Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 21, 2024, at 7:42 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear ChimeraX Help
We made figures using ChimeraX of the same protein. One structure with small molecule bound, the other one without. The cartoon style of the same region is shown differently in these two structures. How can we make them more consistent with each other? The cartoon style width is the same. <image001.png>
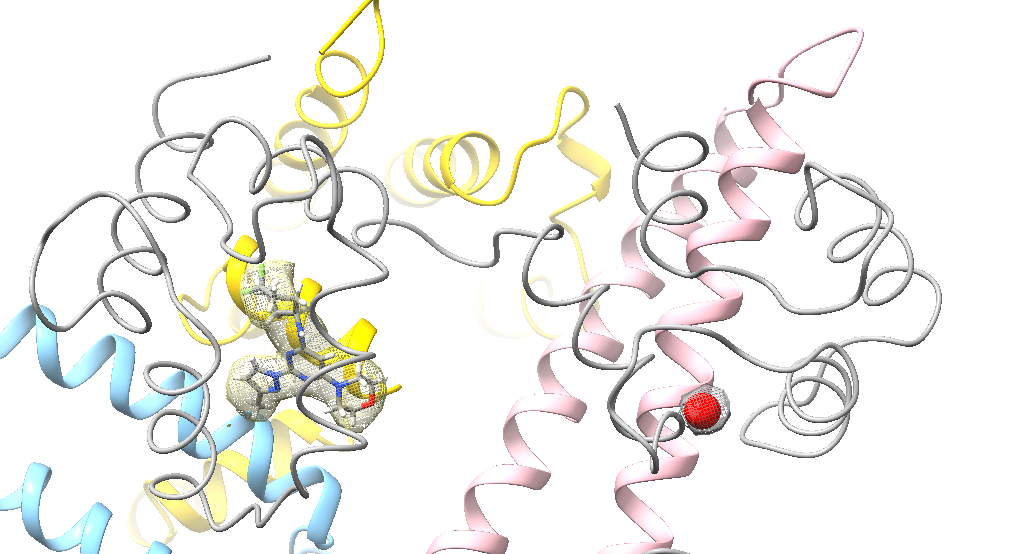
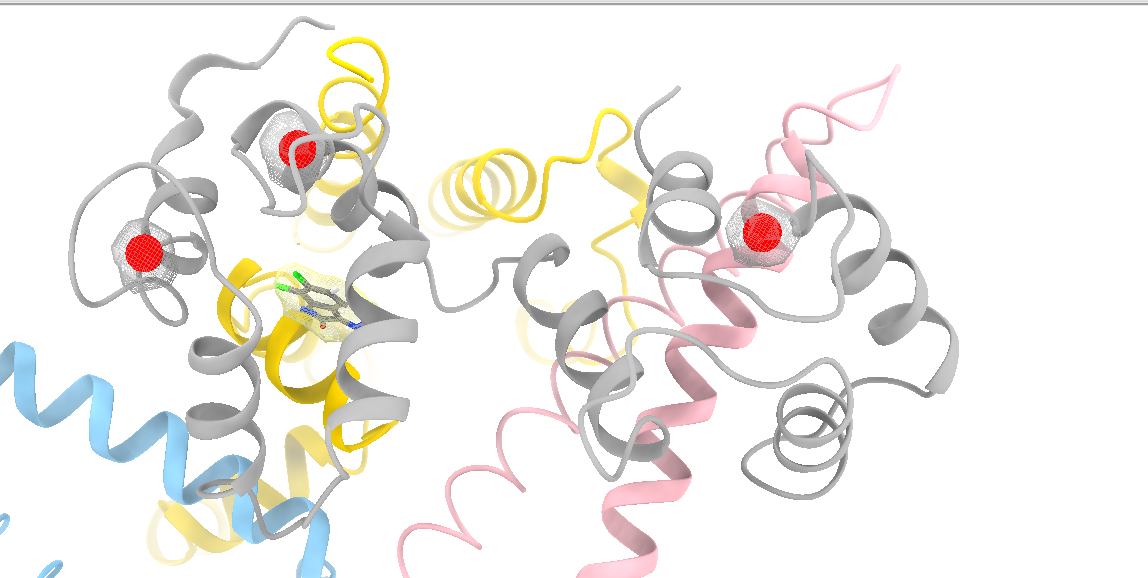
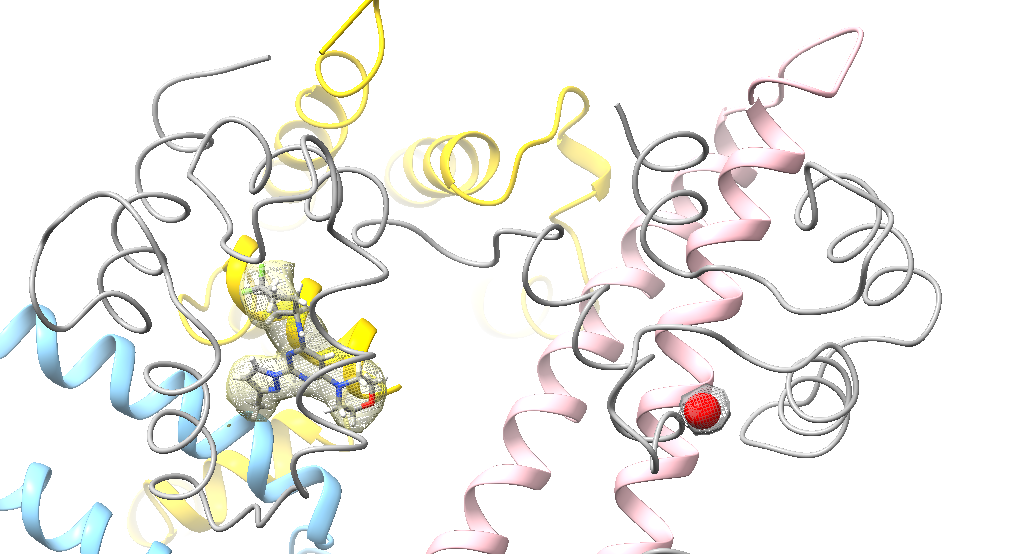
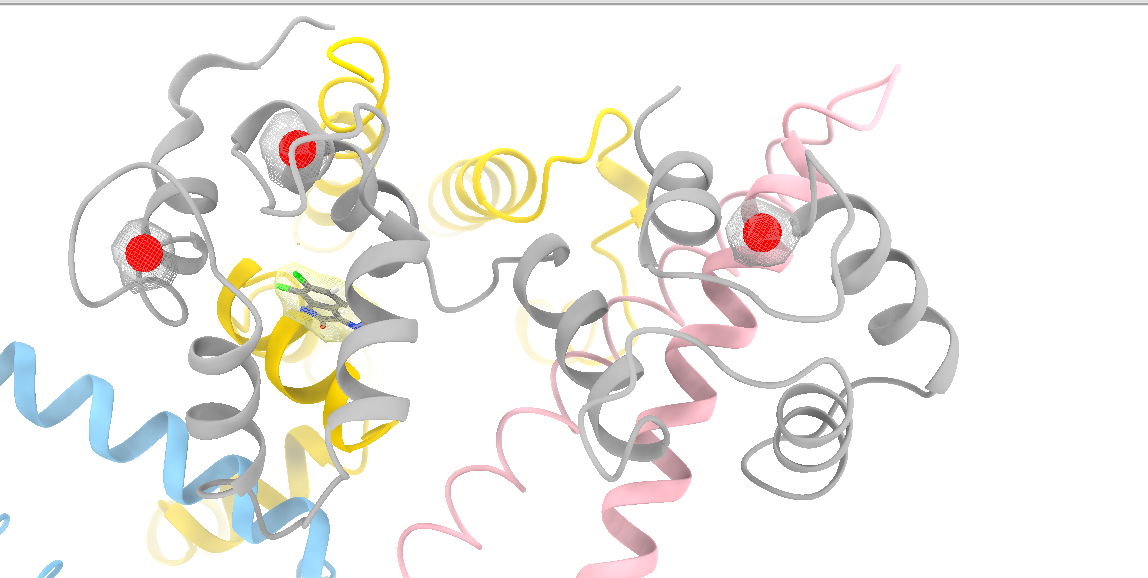
Dear Elaine I read an old post in 2010 about the surfnet tool in Chimera. Is there anything similar to surfnet tool in ChimeraX? CASTp did not find the correct pocket in out apo protein. We want to refine manually and then measure the pocket. Miao From: Elaine Meng <meng@cgl.ucsf.edu> Date: Friday, October 27, 2023 at 1:08 PM To: Zhang, Miao <zhang@chapman.edu> Cc: Tom Goddard via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Subject: change label size External Message [External Message] Hi Miao, If you just want to make current labels bigger, either (A) menu: Actions... Label.. Set Label Height - or - (B) the "label" command "height" option, e.g. label height 2 (or whatever angstrom height you want). For details, see: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Flabel.html&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=QEckL4Ww%2FgF0156cqf7N%2FOJ3lWa3VXY7c%2FTBt%2FMkD%2BI%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html>> Or if you want to set a preference for labels created in the future, see "label defaultHeight" <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fcommands%2Flabel.html%23defaultHeight&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=OMC1nNqy9ywqXAdyv1Kkpl6t%2BDTNzlPP1pnNI1JlnO4%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/commands/label.html#defaultHeight>> ...or, you can do the same thing with the Preferences GUI, the Labels section: <https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Frbvi.ucsf.edu%2Fchimerax%2Fdocs%2Fuser%2Fpreferences.html%23labels&data=05%7C01%7Czhang%40chapman.edu%7Cdb227fbe283641aaecbe08dbd7287e44%7C809929af2d2545bf9837089eb9cfbd01%7C1%7C0%7C638340341143597152%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=ey2EniLYdw9oSelNDM0%2Bmcf7jxJWJoe0aZcvkfs63ws%3D&reserved=0<https://rbvi.ucsf.edu/chimerax/docs/user/preferences.html#labels>> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 27, 2023, at 12:41 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
How do I change the size of label “5.6 angstrom” of distance? Miao
- NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.
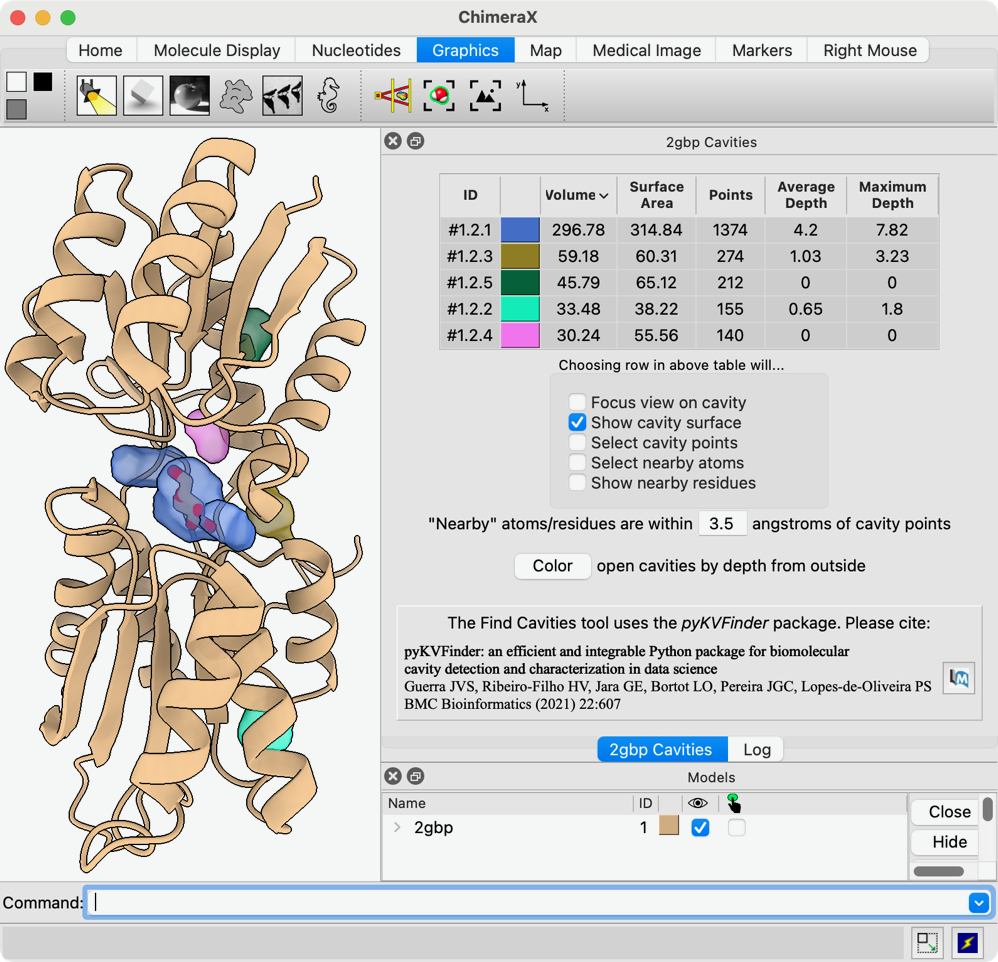
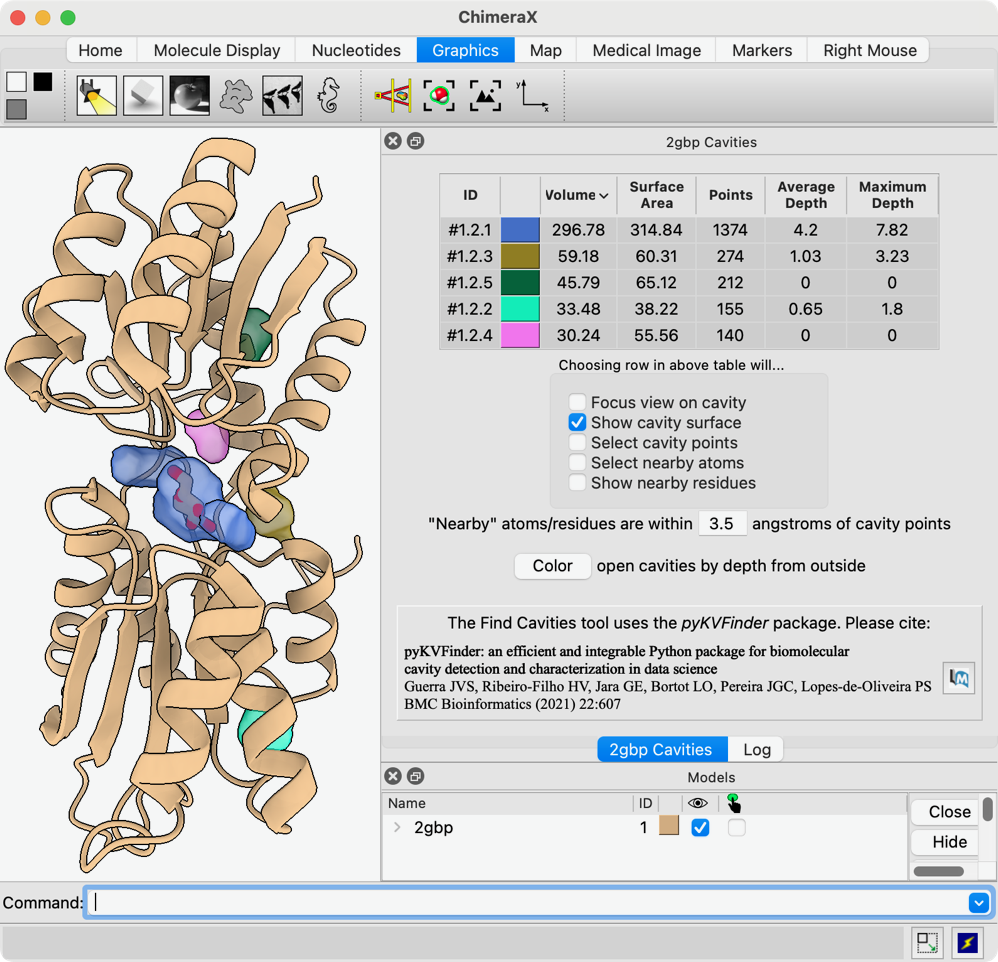
Hi Miao, You have good timing! We have a fairly new tool Find Cavities (in menu under Tools... Structure Analysis) and command "kvfinder" implementing the previously published KVFinder program from the Brazilian Biosciences National Lab. I don't know if the default settings would find your pocket, but you can adjust the probe radii and other parameters as described in the papers and (briefly) in the help pages. Not all of the features described in the help pages made it into the 1.9 release, so you might want to get a newer 1.10 daily build instead to match this documentation: <https://www.rbvi.ucsf.edu/chimerax/docs/user/tools/findcavities.html> <https://www.rbvi.ucsf.edu/chimerax/docs/user/commands/kvfinder.html> Running it gives a list of cavities, their volumes, etc. and displays them as dots and/or surfaces. See screenshot attached. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 30, 2025, at 10:58 AM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Elaine I read an old post in 2010 about the surfnet tool in Chimera. Is there anything similar to surfnet tool in ChimeraX? CASTp did not find the correct pocket in out apo protein. We want to refine manually and then measure the pocket. Miao
{kind=link}
Thanks. That worked. How do I remove the cavities that I am not interested in, when making figure? Miao From: Elaine Meng <meng@cgl.ucsf.edu> Date: Thursday, January 30, 2025 at 12:09 To: Zhang, Miao <zhang@chapman.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: finding protein pockets and tunnels External Message Hi Miao, You have good timing! We have a fairly new tool Find Cavities (in menu under Tools... Structure Analysis) and command "kvfinder" implementing the previously published KVFinder program from the Brazilian Biosciences National Lab. I don't know if the default settings would find your pocket, but you can adjust the probe radii and other parameters as described in the papers and (briefly) in the help pages. Not all of the features described in the help pages made it into the 1.9 release, so you might want to get a newer 1.10 daily build instead to match this documentation: <https://www.rbvi.ucsf.edu/chimerax/docs/user/tools/findcavities.html> <https://www.rbvi.ucsf.edu/chimerax/docs/user/commands/kvfinder.html> Running it gives a list of cavities, their volumes, etc. and displays them as dots and/or surfaces. See screenshot attached. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 30, 2025, at 10:58 AM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Elaine I read an old post in 2010 about the surfnet tool in Chimera. Is there anything similar to surfnet tool in ChimeraX? CASTp did not find the correct pocket in out apo protein. We want to refine manually and then measure the pocket. Miao
NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe. [cid:d621d971-a9c4-4b36-a608-d82f561eca72@namprd11.prod.outlook.com]
{kind=link}
HI Miao, Depending on the options you checked in the tool, it should only display the pockets for the rows you highlighted with the mouse in the pocket list dialog. So it should be easy to just hide the ones you don't want to see by only highlighting the row(s) in the list for the one(s) you want to see. This is described in the help: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/findcavities.html#cavitylist> However if you really want to totally delete the other pockets, look in the Models list (you may need to click the disclosure triangle to expand to sub-models). Each set of dots for one cavity is one model. You can just close all the models for the pockets you don't care about, using the Close button in the Model Panel or with the "close" command. <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelpanel.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/close.html> Either way it would be a good idea to save a session so that if you need to re-make the figure later, it will be easy to restore the status/data. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 30, 2025, at 4:24 PM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks. That worked. How do I remove the cavities that I am not interested in, when making figure? Miao From: Elaine Meng <meng@cgl.ucsf.edu> Date: Thursday, January 30, 2025 at 12:09 To: Zhang, Miao <zhang@chapman.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: finding protein pockets and tunnels External Message
Hi Miao, You have good timing! We have a fairly new tool Find Cavities (in menu under Tools... Structure Analysis) and command "kvfinder" implementing the previously published KVFinder program from the Brazilian Biosciences National Lab.
I don't know if the default settings would find your pocket, but you can adjust the probe radii and other parameters as described in the papers and (briefly) in the help pages. Not all of the features described in the help pages made it into the 1.9 release, so you might want to get a newer 1.10 daily build instead to match this documentation:
<https://www.rbvi.ucsf.edu/chimerax/docs/user/tools/findcavities.html> <https://www.rbvi.ucsf.edu/chimerax/docs/user/commands/kvfinder.html>
Running it gives a list of cavities, their volumes, etc. and displays them as dots and/or surfaces. See screenshot attached.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 30, 2025, at 10:58 AM, Zhang, Miao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Elaine I read an old post in 2010 about the surfnet tool in Chimera. Is there anything similar to surfnet tool in ChimeraX? CASTp did not find the correct pocket in out apo protein. We want to refine manually and then measure the pocket. Miao
NOTE: This email originated from outside Chapman’s network. Do not click links or open attachments unless you recognize the sender and know content is safe.<kvf-highlight.png> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
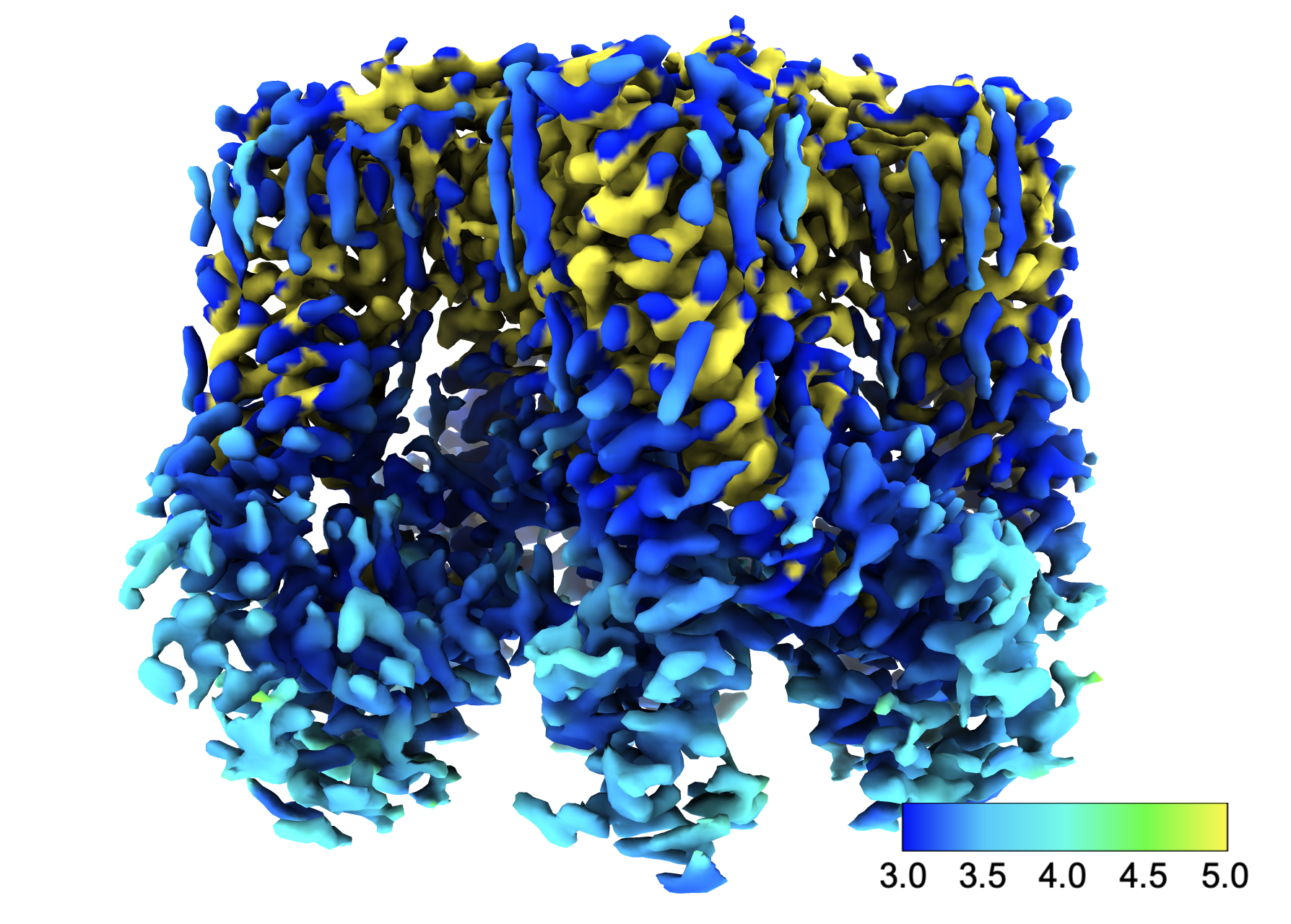
Dear Elaine I tried to use Surface Color to show the local resolution. Please see attached. But something seems odd about the yellow color. The well-defined potion of the map is shown as yellow (5 angstrom). That does not make sense. Is there anything that can cause this kind of mis-coloring? Best, Miao
{kind=link}
No, I don't know of any specific reason for wrong coloring. All I can say is to make sure you are really coloring by the data values that you think you are, (e.g. the map of local resolution values, not by its gradient or something like that), and also that the local resolution map is the one that goes with the map displayed as an isosurface, and is not displaced somehow from that isosurface map (e.g. not moved separately by mistake). Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 26, 2025, at 5:02 PM, Zhang, Miao <zhang@chapman.edu> wrote:
Dear Elaine I tried to use Surface Color to show the local resolution. Please see attached. But something seems odd about the yellow color. The well-defined potion of the map is shown as yellow (5 angstrom). That does not make sense. Is there anything that can cause this kind of mis-coloring? Best, Miao<image1.png>
participants (2)
-
 Elaine Meng
Elaine Meng -
 Zhang, Miao
Zhang, Miao