Colouring Amino Acids Within A Selected Region

Hello ChimeraX developpers, I hope you are doing well. I was trying to colour each amino acid present within a selected region of my model and couldn't figure out how to do it. Could you please guide me on how to do it? I ran these commands (for examples for chains A and E) and have attached my model in this email as well: interfaces select /A contacting /E bothSides true color sel red Please let me know at your earliest convenience. Thank you, Nusrat Tazkia

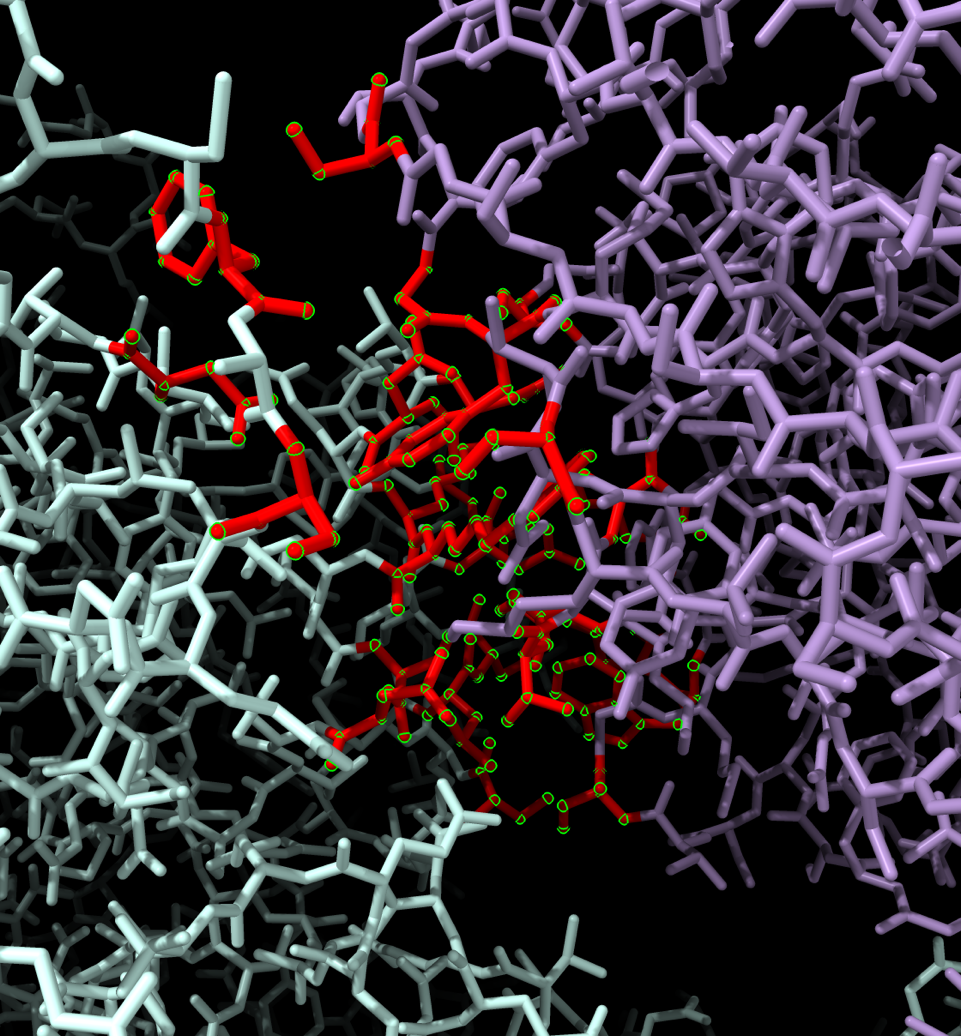
Hi Nusrat, it depends on how you want to define a contact -- chain-chain buried surface area (use "interfaces") ? ...or atom-atom distance (use "contacts")? It might be easier for you to use the menu, Select... Contacts. Then you can choose the Chains tab if you only care about protein or nucleic acid chain-chain contacts (not ligands etc.). If you care about those other kinds of molecules, choose the Atomic tab. There are additional choices for parameter values in those tabs. See the help and links within: <https://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html <mhttps://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html>> Whatever you choose, when you click OK to do the calculation, the resulting command will be generated and shown in the Log. You can use that as an example if you want to use commands directly later. When I tried your molecule and Chains contacts between /A and /E (both chains), buried area >= 15 square angstroms, I got 26 residues selected, see image attached below after coloring them red, changing style to sticks, and zooming in on the selection. The Log showed that the command created by using the menu was: interfaces select /A & ::polymer_type>0 contacting /E & ::polymer_type>0 areaCutoff 0 bothSides true I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 2, 2025, at 10:32 AM, Nusrat Tazkia via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello ChimeraX developpers,
I hope you are doing well. I was trying to colour each amino acid present within a selected region of my model and couldn't figure out how to do it. Could you please guide me on how to do it? I ran these commands (for examples for chains A and E) and have attached my model in this email as well:
interfaces select /A contacting /E bothSides true color sel red
Please let me know at your earliest convenience.
Thank you,
Nusrat Tazkia<palchimera1_model.cif>_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
Actually your command works fine too, gives the same result (26 residues selected, says the Log). What was the problem? Maybe you couldn't see the selection when the atoms were in sphere style? Elaine "interfaces" help <https://rbvi.ucsf.edu/chimerax/docs/user/commands/interfaces.html>
On Dec 2, 2025, at 11:58 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Nusrat, it depends on how you want to define a contact -- chain-chain buried surface area (use "interfaces") ? ...or atom-atom distance (use "contacts")?
It might be easier for you to use the menu, Select... Contacts. Then you can choose the Chains tab if you only care about protein or nucleic acid chain-chain contacts (not ligands etc.). If you care about those other kinds of molecules, choose the Atomic tab. There are additional choices for parameter values in those tabs.
See the help and links within: <https://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html <mhttps://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html>>
Whatever you choose, when you click OK to do the calculation, the resulting command will be generated and shown in the Log. You can use that as an example if you want to use commands directly later.
When I tried your molecule and Chains contacts between /A and /E (both chains), buried area >= 15 square angstroms, I got 26 residues selected, see image attached below after coloring them red, changing style to sticks, and zooming in on the selection.
The Log showed that the command created by using the menu was:
interfaces select /A & ::polymer_type>0 contacting /E & ::polymer_type>0 areaCutoff 0 bothSides true
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
<contacts.png>
On Dec 2, 2025, at 10:32 AM, Nusrat Tazkia via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello ChimeraX developpers,
I hope you are doing well. I was trying to colour each amino acid present within a selected region of my model and couldn't figure out how to do it. Could you please guide me on how to do it? I ran these commands (for examples for chains A and E) and have attached my model in this email as well:
interfaces select /A contacting /E bothSides true color sel red
Please let me know at your earliest convenience.
Thank you,
Nusrat Tazkia<palchimera1_model.cif>_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hi Elaine, Yeah, so I just wanted to colour the amino acids within those 26 residues with different colours for each of them. Right now, I can only colour the entire selection using "color sel red" and I wasn't sure how to colour each amino acid within that selection to have a different colour. Please let me know how I can do that. Thank you, Nusrat Tazkia ________________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Tuesday, December 2, 2025 3:06 PM To: Nusrat Tazkia <ntazk024@uottawa.ca> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Colouring Amino Acids Within A Selected Region Attention : courriel externe | external email Actually your command works fine too, gives the same result (26 residues selected, says the Log). What was the problem? Maybe you couldn't see the selection when the atoms were in sphere style? Elaine "interfaces" help <https://rbvi.ucsf.edu/chimerax/docs/user/commands/interfaces.html>
On Dec 2, 2025, at 11:58 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Nusrat, it depends on how you want to define a contact -- chain-chain buried surface area (use "interfaces") ? ...or atom-atom distance (use "contacts")?
It might be easier for you to use the menu, Select... Contacts. Then you can choose the Chains tab if you only care about protein or nucleic acid chain-chain contacts (not ligands etc.). If you care about those other kinds of molecules, choose the Atomic tab. There are additional choices for parameter values in those tabs.
See the help and links within: <https://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html <mhttps://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html>>
Whatever you choose, when you click OK to do the calculation, the resulting command will be generated and shown in the Log. You can use that as an example if you want to use commands directly later.
When I tried your molecule and Chains contacts between /A and /E (both chains), buried area >= 15 square angstroms, I got 26 residues selected, see image attached below after coloring them red, changing style to sticks, and zooming in on the selection.
The Log showed that the command created by using the menu was:
interfaces select /A & ::polymer_type>0 contacting /E & ::polymer_type>0 areaCutoff 0 bothSides true
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
<contacts.png>
On Dec 2, 2025, at 10:32 AM, Nusrat Tazkia via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello ChimeraX developpers,
I hope you are doing well. I was trying to colour each amino acid present within a selected region of my model and couldn't figure out how to do it. Could you please guide me on how to do it? I ran these commands (for examples for chains A and E) and have attached my model in this email as well:
interfaces select /A contacting /E bothSides true color sel red
Please let me know at your earliest convenience.
Thank you,
Nusrat Tazkia<palchimera1_model.cif>_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hi Nusrat, Yes, please ask the question precisely to avoid wasting time. Your commands did color all your selected residues, it was the "differently" part that was not clear. To color a selection so that each residue in the selection has a different color, you could try color sel random However, that colors randomly *per atom*, so there will be multiple colors within a single residue. To color a selection of residues so that each residue has a different color, you could try "rainbow" ... however, the colors of multiple residues will be similar to each other since it gradually changes the color across the palette. I.e. there will be many greenish residues only a slightly different color of green, if when your 26 residues are selected, if you use command: rainbow sel ... which uses default palette of rainbow colors. You could try using a palette with more different colors, rainbow sel paired-12 .... but it will still have the same issue for a selection of 26 residues,where some residues will have similar colors even though they are slightly different. To fully control the residue coloring you would need to do it the long way, list in the Log the residue numbers in the selection (e.g. "info residues sel") and then manually color each one by one using a specified color (e.g. "color /E:199 dodger blue") See help pages: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/color.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/colornames.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/palettes.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 3, 2025, at 6:52 AM, Nusrat Tazkia via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Yeah, so I just wanted to colour the amino acids within those 26 residues with different colours for each of them. Right now, I can only colour the entire selection using "color sel red" and I wasn't sure how to colour each amino acid within that selection to have a different colour. Please let me know how I can do that.
Thank you,
Nusrat Tazkia
________________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Tuesday, December 2, 2025 3:06 PM To: Nusrat Tazkia <ntazk024@uottawa.ca> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Colouring Amino Acids Within A Selected Region
Attention : courriel externe | external email
Actually your command works fine too, gives the same result (26 residues selected, says the Log).
What was the problem? Maybe you couldn't see the selection when the atoms were in sphere style?
Elaine
"interfaces" help <https://rbvi.ucsf.edu/chimerax/docs/user/commands/interfaces.html>
On Dec 2, 2025, at 11:58 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Nusrat, it depends on how you want to define a contact -- chain-chain buried surface area (use "interfaces") ? ...or atom-atom distance (use "contacts")?
It might be easier for you to use the menu, Select... Contacts. Then you can choose the Chains tab if you only care about protein or nucleic acid chain-chain contacts (not ligands etc.). If you care about those other kinds of molecules, choose the Atomic tab. There are additional choices for parameter values in those tabs.
See the help and links within: <https://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html <mhttps://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html>>
Whatever you choose, when you click OK to do the calculation, the resulting command will be generated and shown in the Log. You can use that as an example if you want to use commands directly later.
When I tried your molecule and Chains contacts between /A and /E (both chains), buried area >= 15 square angstroms, I got 26 residues selected, see image attached below after coloring them red, changing style to sticks, and zooming in on the selection.
The Log showed that the command created by using the menu was:
interfaces select /A & ::polymer_type>0 contacting /E & ::polymer_type>0 areaCutoff 0 bothSides true
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
<contacts.png>
On Dec 2, 2025, at 10:32 AM, Nusrat Tazkia via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello ChimeraX developpers,
I hope you are doing well. I was trying to colour each amino acid present within a selected region of my model and couldn't figure out how to do it. Could you please guide me on how to do it? I ran these commands (for examples for chains A and E) and have attached my model in this email as well:
interfaces select /A contacting /E bothSides true color sel red
Please let me know at your earliest convenience.
Thank you,
Nusrat Tazkia<palchimera1_model.cif>
Hi Elaine, I see. Thank you so much for your detailed answer! Thank you, Nusrat Tazkia ________________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, December 3, 2025 12:13 PM To: Nusrat Tazkia <ntazk024@uottawa.ca> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Colouring Amino Acids Within A Selected Region Attention : courriel externe | external email Hi Nusrat, Yes, please ask the question precisely to avoid wasting time. Your commands did color all your selected residues, it was the "differently" part that was not clear. To color a selection so that each residue in the selection has a different color, you could try color sel random However, that colors randomly *per atom*, so there will be multiple colors within a single residue. To color a selection of residues so that each residue has a different color, you could try "rainbow" ... however, the colors of multiple residues will be similar to each other since it gradually changes the color across the palette. I.e. there will be many greenish residues only a slightly different color of green, if when your 26 residues are selected, if you use command: rainbow sel ... which uses default palette of rainbow colors. You could try using a palette with more different colors, rainbow sel paired-12 .... but it will still have the same issue for a selection of 26 residues,where some residues will have similar colors even though they are slightly different. To fully control the residue coloring you would need to do it the long way, list in the Log the residue numbers in the selection (e.g. "info residues sel") and then manually color each one by one using a specified color (e.g. "color /E:199 dodger blue") See help pages: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/color.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/colornames.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/palettes.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 3, 2025, at 6:52 AM, Nusrat Tazkia via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Yeah, so I just wanted to colour the amino acids within those 26 residues with different colours for each of them. Right now, I can only colour the entire selection using "color sel red" and I wasn't sure how to colour each amino acid within that selection to have a different colour. Please let me know how I can do that.
Thank you,
Nusrat Tazkia
________________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Tuesday, December 2, 2025 3:06 PM To: Nusrat Tazkia <ntazk024@uottawa.ca> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Colouring Amino Acids Within A Selected Region
Attention : courriel externe | external email
Actually your command works fine too, gives the same result (26 residues selected, says the Log).
What was the problem? Maybe you couldn't see the selection when the atoms were in sphere style?
Elaine
"interfaces" help <https://rbvi.ucsf.edu/chimerax/docs/user/commands/interfaces.html>
On Dec 2, 2025, at 11:58 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Nusrat, it depends on how you want to define a contact -- chain-chain buried surface area (use "interfaces") ? ...or atom-atom distance (use "contacts")?
It might be easier for you to use the menu, Select... Contacts. Then you can choose the Chains tab if you only care about protein or nucleic acid chain-chain contacts (not ligands etc.). If you care about those other kinds of molecules, choose the Atomic tab. There are additional choices for parameter values in those tabs.
See the help and links within: <https://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html <mhttps://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html>>
Whatever you choose, when you click OK to do the calculation, the resulting command will be generated and shown in the Log. You can use that as an example if you want to use commands directly later.
When I tried your molecule and Chains contacts between /A and /E (both chains), buried area >= 15 square angstroms, I got 26 residues selected, see image attached below after coloring them red, changing style to sticks, and zooming in on the selection.
The Log showed that the command created by using the menu was:
interfaces select /A & ::polymer_type>0 contacting /E & ::polymer_type>0 areaCutoff 0 bothSides true
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
<contacts.png>
On Dec 2, 2025, at 10:32 AM, Nusrat Tazkia via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello ChimeraX developpers,
I hope you are doing well. I was trying to colour each amino acid present within a selected region of my model and couldn't figure out how to do it. Could you please guide me on how to do it? I ran these commands (for examples for chains A and E) and have attached my model in this email as well:
interfaces select /A contacting /E bothSides true color sel red
Please let me know at your earliest convenience.
Thank you,
Nusrat Tazkia<palchimera1_model.cif>
participants (2)
-
 Elaine Meng
Elaine Meng -
 Nusrat Tazkia
Nusrat Tazkia