Re: [chimerax-users] Assembling 2 proteins

Hi Tata Santosh Rama Bhadra Rao, I already gave my suggestions in the previous message, so there is not really anything else I can do to teach you. Since you didn't give the details of all the steps that you tried and which step(s) gave results you didn't like, I can't tell you how to do it differently. My only idea is to go back to what I already suggested: you could use Matchmaker if you already had a pentamer, which you show in the bottom figures. So, following the instructions in my first reply, maybe you can take the chains shown in the top figures and match them onto the chains in the bottom figures to make the pentamer. However, that will only work if the proteins are very similar. If you did it already and it gave the bad result, maybe these proteins are not similar enough to use that approach. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On May 30, 2022, at 9:22 PM, SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> wrote:
Hi Elaine,
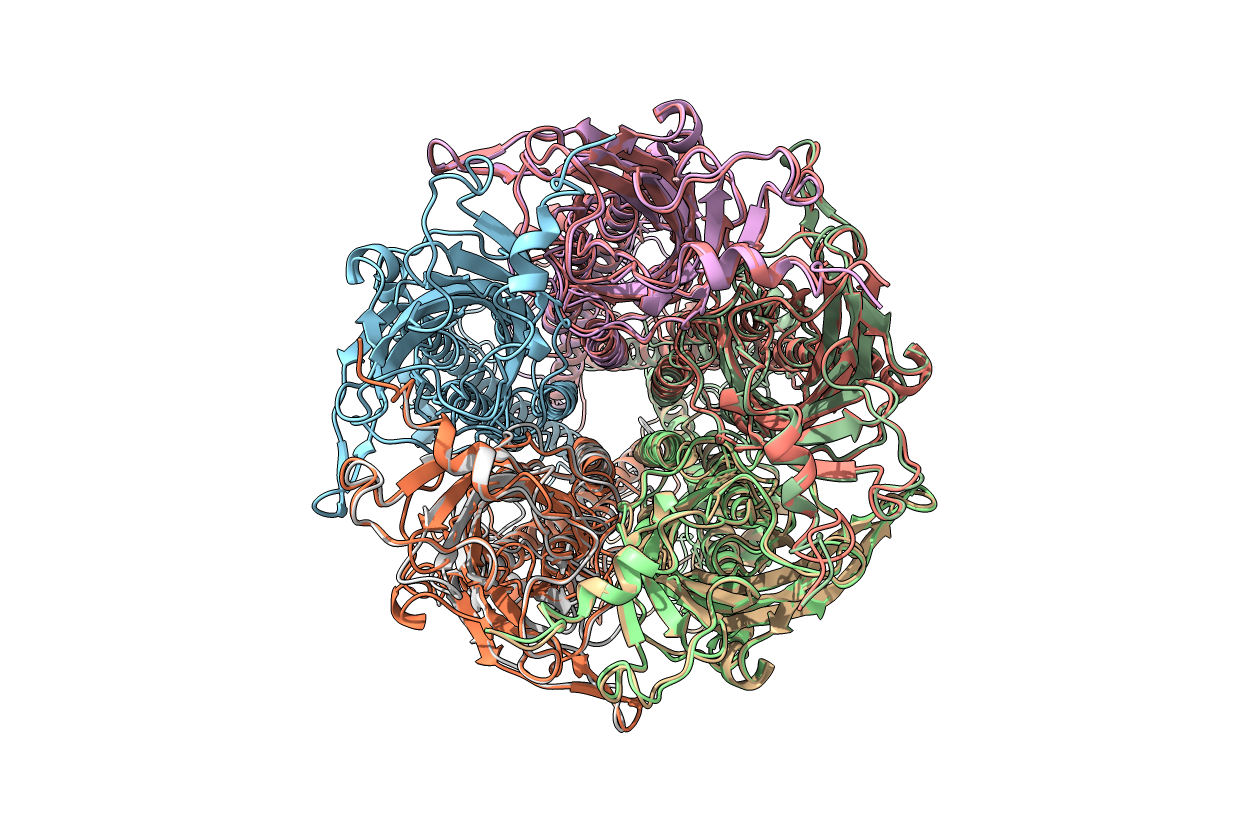
I tried doing and it came out like this. Final 2 figures are homomeric pentamers. First 2 are heteromers with 3A subunits (A, B, D chains) and 2D (C, E chains) subunits. Could you please help me or teach me assembling them properly.
Thanking you,
With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974 From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, 25 May 2022 2:08 AM To: SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Assembling 2 proteins
Hello Tata Santosh Rama Bhadra Rao, It all depends on what information you have already. If you don't know how the pentamer is supposed to look (how the proteins fit together), then there may not be enough information: ChimeraX is not going to predict the pentamer structure for you.
You said "5 different proteins" which I interpret to mean they are not 5 copies of the same thing, that it is a heteropentamer and not a homopentamer. If they were 5 copies of the same thing (homopentamer), then you might be able to use the "sym" command to build it from one copy, if the input file contains symmetry matrices in the header, or you know the symmetry and can specify it separately: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/sym.html>
Otherwise: You would only use Matchmaker if you also have an already-known structure of a similar pentamer. Then you would open the structure of that pentamer, open the structures of your five proteins (if the same one is multiple copies in the pentamer, you would open it multiple times), and then match each one of them to the similar subunit of the known pentamer. In other words, you would open 6 different PDB files and then use Matchmaker 5 times, once to match each of your proteins to the corresponding subunit of the pentamer.
Just start Matchmaker (menu: Tools... Structure Comparison... Matchmaker) and in that dialog's Chain Pairing, choose "Specific chain(s) in reference structure with specific chain(s) in match structure". Choose one of your protein chains as the match structure and choose the corresponding chain in the known pentamer as the reference. Apply. Repeat with your second protein chain, etc.
Here is the Matchmaker help with detailed explanations of all the options: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/matchmaker.html#top>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On May 23, 2022, at 9:49 PM, SANTOSH RAMA BHADRA RAO TATA via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi,
I would like to create a pentamer, how can I assemble 5 different proteins subunits into a pentamer. I was trying to use matchmaker but I couldn't. can you please help me with some video how to get in use it or if there is any other way to use the software and create pentamer please let me know.
Thanking you,
With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974

Dear Meng, It worked thank you very much for your suggestions. I was able to create the pentamer. Thanking you, With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974 ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, 1 June 2022 1:50 AM To: SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Assembling 2 proteins Hi Tata Santosh Rama Bhadra Rao, I already gave my suggestions in the previous message, so there is not really anything else I can do to teach you. Since you didn't give the details of all the steps that you tried and which step(s) gave results you didn't like, I can't tell you how to do it differently. My only idea is to go back to what I already suggested: you could use Matchmaker if you already had a pentamer, which you show in the bottom figures. So, following the instructions in my first reply, maybe you can take the chains shown in the top figures and match them onto the chains in the bottom figures to make the pentamer. However, that will only work if the proteins are very similar. If you did it already and it gave the bad result, maybe these proteins are not similar enough to use that approach. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On May 30, 2022, at 9:22 PM, SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> wrote:
Hi Elaine,
I tried doing and it came out like this. Final 2 figures are homomeric pentamers. First 2 are heteromers with 3A subunits (A, B, D chains) and 2D (C, E chains) subunits. Could you please help me or teach me assembling them properly.
Thanking you,
With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974 From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, 25 May 2022 2:08 AM To: SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Assembling 2 proteins
Hello Tata Santosh Rama Bhadra Rao, It all depends on what information you have already. If you don't know how the pentamer is supposed to look (how the proteins fit together), then there may not be enough information: ChimeraX is not going to predict the pentamer structure for you.
You said "5 different proteins" which I interpret to mean they are not 5 copies of the same thing, that it is a heteropentamer and not a homopentamer. If they were 5 copies of the same thing (homopentamer), then you might be able to use the "sym" command to build it from one copy, if the input file contains symmetry matrices in the header, or you know the symmetry and can specify it separately: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/sym.html<https://rbvi.ucsf.edu/chimerax/docs/user/commands/sym.html>>
Otherwise: You would only use Matchmaker if you also have an already-known structure of a similar pentamer. Then you would open the structure of that pentamer, open the structures of your five proteins (if the same one is multiple copies in the pentamer, you would open it multiple times), and then match each one of them to the similar subunit of the known pentamer. In other words, you would open 6 different PDB files and then use Matchmaker 5 times, once to match each of your proteins to the corresponding subunit of the pentamer.
Just start Matchmaker (menu: Tools... Structure Comparison... Matchmaker) and in that dialog's Chain Pairing, choose "Specific chain(s) in reference structure with specific chain(s) in match structure". Choose one of your protein chains as the match structure and choose the corresponding chain in the known pentamer as the reference. Apply. Repeat with your second protein chain, etc.
Here is the Matchmaker help with detailed explanations of all the options: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/matchmaker.html#top<https://rbvi.ucsf.edu/chimerax/docs/user/tools/matchmaker.html#top>>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On May 23, 2022, at 9:49 PM, SANTOSH RAMA BHADRA RAO TATA via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi,
I would like to create a pentamer, how can I assemble 5 different proteins subunits into a pentamer. I was trying to use matchmaker but I couldn't. can you please help me with some video how to get in use it or if there is any other way to use the software and create pentamer please let me know.
Thanking you,
With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974
{kind=link}
Good day Meng, I'm trying to find the tutorial for the measuring the pore diameter. Can you please help me which protocol goes well with it. Thanking you, With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974 ________________________________ From: SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> Sent: Monday, 18 July 2022 2:32 PM To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Assembling 2 proteins Dear Meng, It worked thank you very much for your suggestions. I was able to create the pentamer. Thanking you, With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974 ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, 1 June 2022 1:50 AM To: SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Assembling 2 proteins Hi Tata Santosh Rama Bhadra Rao, I already gave my suggestions in the previous message, so there is not really anything else I can do to teach you. Since you didn't give the details of all the steps that you tried and which step(s) gave results you didn't like, I can't tell you how to do it differently. My only idea is to go back to what I already suggested: you could use Matchmaker if you already had a pentamer, which you show in the bottom figures. So, following the instructions in my first reply, maybe you can take the chains shown in the top figures and match them onto the chains in the bottom figures to make the pentamer. However, that will only work if the proteins are very similar. If you did it already and it gave the bad result, maybe these proteins are not similar enough to use that approach. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On May 30, 2022, at 9:22 PM, SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> wrote:
Hi Elaine,
I tried doing and it came out like this. Final 2 figures are homomeric pentamers. First 2 are heteromers with 3A subunits (A, B, D chains) and 2D (C, E chains) subunits. Could you please help me or teach me assembling them properly.
Thanking you,
With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974 From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, 25 May 2022 2:08 AM To: SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Assembling 2 proteins
Hello Tata Santosh Rama Bhadra Rao, It all depends on what information you have already. If you don't know how the pentamer is supposed to look (how the proteins fit together), then there may not be enough information: ChimeraX is not going to predict the pentamer structure for you.
You said "5 different proteins" which I interpret to mean they are not 5 copies of the same thing, that it is a heteropentamer and not a homopentamer. If they were 5 copies of the same thing (homopentamer), then you might be able to use the "sym" command to build it from one copy, if the input file contains symmetry matrices in the header, or you know the symmetry and can specify it separately: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/sym.html<https://rbvi.ucsf.edu/chimerax/docs/user/commands/sym.html>>
Otherwise: You would only use Matchmaker if you also have an already-known structure of a similar pentamer. Then you would open the structure of that pentamer, open the structures of your five proteins (if the same one is multiple copies in the pentamer, you would open it multiple times), and then match each one of them to the similar subunit of the known pentamer. In other words, you would open 6 different PDB files and then use Matchmaker 5 times, once to match each of your proteins to the corresponding subunit of the pentamer.
Just start Matchmaker (menu: Tools... Structure Comparison... Matchmaker) and in that dialog's Chain Pairing, choose "Specific chain(s) in reference structure with specific chain(s) in match structure". Choose one of your protein chains as the match structure and choose the corresponding chain in the known pentamer as the reference. Apply. Repeat with your second protein chain, etc.
Here is the Matchmaker help with detailed explanations of all the options: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/matchmaker.html#top<https://rbvi.ucsf.edu/chimerax/docs/user/tools/matchmaker.html#top>>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On May 23, 2022, at 9:49 PM, SANTOSH RAMA BHADRA RAO TATA via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi,
I would like to create a pentamer, how can I assemble 5 different proteins subunits into a pentamer. I was trying to use matchmaker but I couldn't. can you please help me with some video how to get in use it or if there is any other way to use the software and create pentamer please let me know.
Thanking you,
With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974
Good day Tata Santosh Rama Bhadra Rao, There is no tutorial for measuring pore diameter, sorry. ChimeraX does not have a tool specifically to show a tunnel and report the pore radius -- if you wanted something like that maybe you would need to use other software like HOLE, MOLE, Caver, etc., maybe MolAxis. For example, here are MOLEonline and MolAxis servers: <https://mole.upol.cz/> <https://bioinfo3d.cs.tau.ac.il/MolAxis/> However, if you are willing to choose two points yourself in ChimeraX, you can measure the distance between them. Some possible approaches: (1) You can measure the distance between any two atoms, e.g. show atoms, Ctrl-click to select first atom, Shift-Ctrl-doubleclick second atom, choose "Distance" from resulting context menu (2) You can use the "tape measure" mouse mode to click and drag a measurement. This can be between atoms, ribbons, and/or surface points. First you have to assign a button to this function and then use it, e.g. command mouse right "tape measure" <https://rbvi.ucsf.edu/chimerax/docs/user/commands/ui.html#mousemode> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/mousemodes.html#tapemeasure> ...and then right click-drag to do the measurements. However, it can be very difficult to get to the positions you want to measure, especially if you are showing a tunnel surface. You may need to use clipping such as with the Side View (menu: Tools... General... Side View) and even then it may not work the way you would like. In that case you would need to use other software as mentioned above. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 26, 2022, at 8:18 PM, SANTOSH RAMA BHADRA RAO TATA via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Good day Meng,
I'm trying to find the tutorial for the measuring the pore diameter. Can you please help me which protocol goes well with it.
Thanking you,
With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974
Good morning Meng, Thank you very much for the response will use the suggestions to get details from it. Thanking you, With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974 ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Thursday, 28 July 2022 2:22 AM To: SANTOSH RAMA BHADRA RAO TATA <19807877@students.latrobe.edu.au> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Measuring pore diameter Good day Tata Santosh Rama Bhadra Rao, There is no tutorial for measuring pore diameter, sorry. ChimeraX does not have a tool specifically to show a tunnel and report the pore radius -- if you wanted something like that maybe you would need to use other software like HOLE, MOLE, Caver, etc., maybe MolAxis. For example, here are MOLEonline and MolAxis servers: <https://mole.upol.cz/<https://mole.upol.cz>> <https://bioinfo3d.cs.tau.ac.il/MolAxis/<https://bioinfo3d.cs.tau.ac.il/MolAxis>> However, if you are willing to choose two points yourself in ChimeraX, you can measure the distance between them. Some possible approaches: (1) You can measure the distance between any two atoms, e.g. show atoms, Ctrl-click to select first atom, Shift-Ctrl-doubleclick second atom, choose "Distance" from resulting context menu (2) You can use the "tape measure" mouse mode to click and drag a measurement. This can be between atoms, ribbons, and/or surface points. First you have to assign a button to this function and then use it, e.g. command mouse right "tape measure" <https://rbvi.ucsf.edu/chimerax/docs/user/commands/ui.html#mousemode<https://rbvi.ucsf.edu/chimerax/docs/user/commands/ui.html#mousemode>> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/mousemodes.html#tapemeasure<https://rbvi.ucsf.edu/chimerax/docs/user/tools/mousemodes.html#tapemeasure>> ...and then right click-drag to do the measurements. However, it can be very difficult to get to the positions you want to measure, especially if you are showing a tunnel surface. You may need to use clipping such as with the Side View (menu: Tools... General... Side View) and even then it may not work the way you would like. In that case you would need to use other software as mentioned above. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 26, 2022, at 8:18 PM, SANTOSH RAMA BHADRA RAO TATA via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Good day Meng,
I'm trying to find the tutorial for the measuring the pore diameter. Can you please help me which protocol goes well with it.
Thanking you,
With best wishes, Tata Santosh Rama Bhadra Rao, PhD Scholar, C/O Prof Helen Irving, La Trobe university, Bendigo, Vic-3550, Australia. Email: 19807877@students.latrobe.edu.au; S.Tata@latrobe.edu.au Phone: 0499263974
participants (2)
-
 Elaine Meng
Elaine Meng -
 SANTOSH RAMA BHADRA RAO TATA
SANTOSH RAMA BHADRA RAO TATA