
Hello, I hope this email finds you well. My name is Louis OIiveira, and I am a Master's student specializing in protein crystallography at the University of São Paulo. I am currently using Chimera X for generating images of the structure I am studying. However, I have encountered some issues that I am struggling to resolve. Specifically, I have successfully validated the structure of the enzyme and am now attempting to present the co-crystallized ligand along with its electronic density map interacting with the enzyme. My challenge lies in isolating the electronic density of the ligand without including the electronic density of the nearby residues. I have attempted to use the "zone" function in the map menu, but I am still seeing some electronic density that I wish to exclude as shown in the picture . I would greatly appreciate your guidance on how to effectively isolate the electronic density of the ligand in Chimera X. If you have any insights or recommendations, I would be grateful for your assistance. Thank you for your time, and I look forward to hearing from you soon. Best regards, Louis Oliveira Master's Student Instituto de Fisica de São Carlos University of São Paulo 
{kind=link}

Hi Louis, You'll see when you use the Zone button in the Map toolbar that it logs a command that looks like volume zone #1 nearAtoms sel & #2 range 9.25 Just copy and paste that command to the command line at the bottom of the ChimeraX window and change the range value to a smaller value, for example, volume zone #1 nearAtoms sel & #2 range 5 The range value is the maximum distance away from the selected ligand atoms to show the density. Documentation for the volume zone command is here https://www.cgl.ucsf.edu/chimerax/docs/user/commands/volume.html#zone Tom
On Dec 15, 2023, at 3:18 PM, Louis Fellipe Moreno Oliveira via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I hope this email finds you well. My name is Louis OIiveira, and I am a Master's student specializing in protein crystallography at the University of São Paulo.
I am currently using Chimera X for generating images of the structure I am studying. However, I have encountered some issues that I am struggling to resolve. Specifically, I have successfully validated the structure of the enzyme and am now attempting to present the co-crystallized ligand along with its electronic density map interacting with the enzyme.
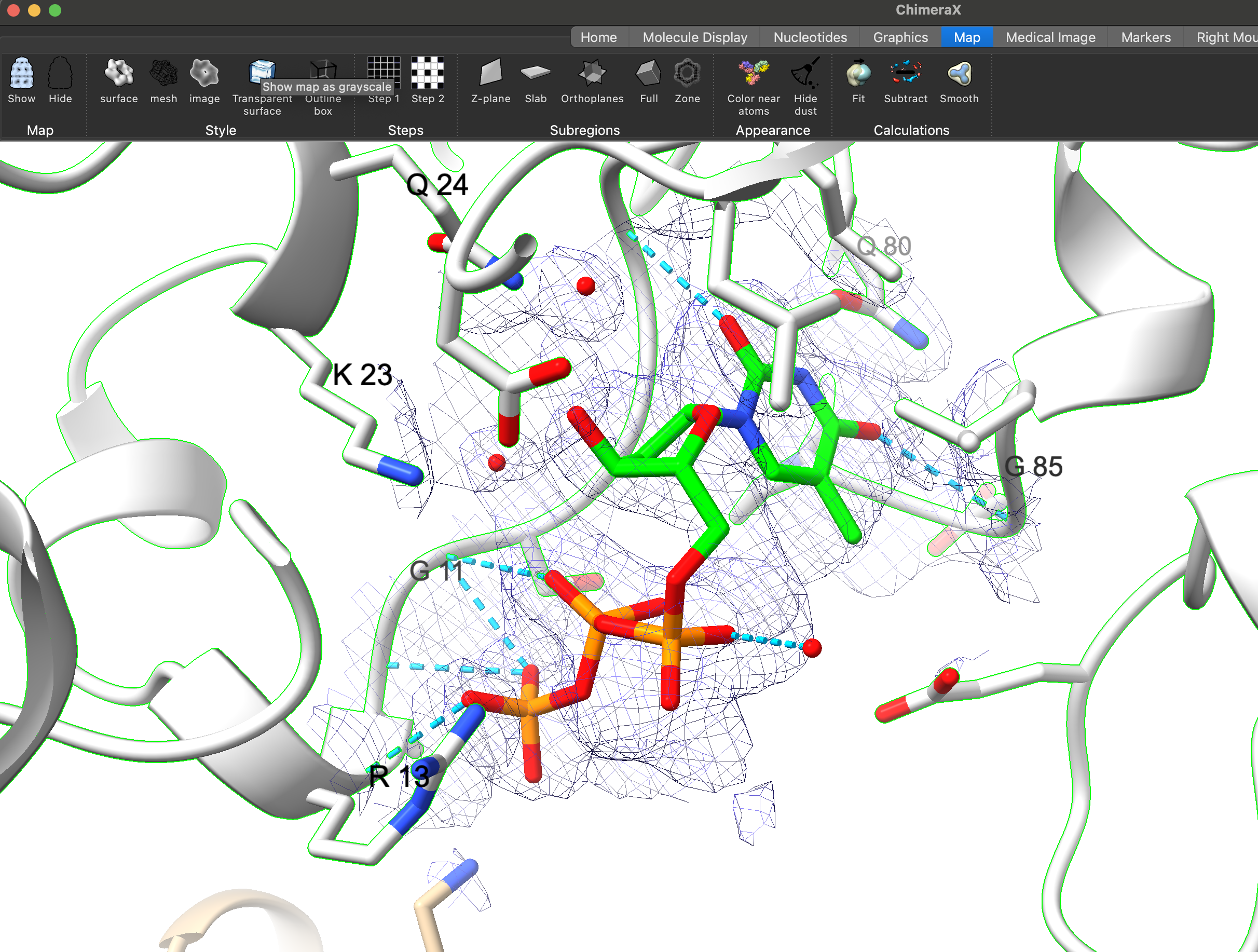
My challenge lies in isolating the electronic density of the ligand without including the electronic density of the nearby residues. I have attempted to use the "zone" function in the map menu, but I am still seeing some electronic density that I wish to exclude as shown in the picture .
I would greatly appreciate your guidance on how to effectively isolate the electronic density of the ligand in Chimera X. If you have any insights or recommendations, I would be grateful for your assistance.
Thank you for your time, and I look forward to hearing from you soon.
Best regards,
Louis Oliveira Master's Student Instituto de Fisica de São Carlos University of São Paulo
<Captura de Tela 2023-12-15 às 20.14.05.png> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Or maybe easier, use the Surface Zone graphical user interface, menu Tools / Volume Data / Surface Zone. That has a slider that you can adjust to control how far away the density is shown. Tom
On Dec 15, 2023, at 5:03 PM, Tom Goddard <goddard@sonic.net> wrote:
Hi Louis,
You'll see when you use the Zone button in the Map toolbar that it logs a command that looks like
volume zone #1 nearAtoms sel & #2 range 9.25
Just copy and paste that command to the command line at the bottom of the ChimeraX window and change the range value to a smaller value, for example,
volume zone #1 nearAtoms sel & #2 range 5
The range value is the maximum distance away from the selected ligand atoms to show the density. Documentation for the volume zone command is here
https://www.cgl.ucsf.edu/chimerax/docs/user/commands/volume.html#zone
Tom
On Dec 15, 2023, at 3:18 PM, Louis Fellipe Moreno Oliveira via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I hope this email finds you well. My name is Louis OIiveira, and I am a Master's student specializing in protein crystallography at the University of São Paulo.
I am currently using Chimera X for generating images of the structure I am studying. However, I have encountered some issues that I am struggling to resolve. Specifically, I have successfully validated the structure of the enzyme and am now attempting to present the co-crystallized ligand along with its electronic density map interacting with the enzyme.
My challenge lies in isolating the electronic density of the ligand without including the electronic density of the nearby residues. I have attempted to use the "zone" function in the map menu, but I am still seeing some electronic density that I wish to exclude as shown in the picture .
I would greatly appreciate your guidance on how to effectively isolate the electronic density of the ligand in Chimera X. If you have any insights or recommendations, I would be grateful for your assistance.
Thank you for your time, and I look forward to hearing from you soon.
Best regards,
Louis Oliveira Master's Student Instituto de Fisica de São Carlos University of São Paulo
<Captura de Tela 2023-12-15 às 20.14.05.png> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
participants (2)
-
 Louis Fellipe Moreno Oliveira
Louis Fellipe Moreno Oliveira -
 Tom Goddard
Tom Goddard