Label “check_interface” residues in ChimeraX

Hello ChimeraX team, I came to know this fabulous ChimeraX custom “check_interface” command to instantly identify interface residues between chains A and B of protein complex model #1. It works nicely with any number of protein complex models for e.g. #1, 2, 3, 4 with chains A and B However, when I am trying to “label” the identified interface residues using the below mentioned ChimeraX command for the various protein complex models #1, 2, 3, 4. It doesn’t seem to work. I am interested to identify and label the interface residues for the related set of various protein complex models #1, 2, 3,4. label sel text "{0.label_one_letter_code}{0.number}" Can somebody suggest what I am doing wrong with the ChimeraX “label” command ?? Thanks a lot ! Arun Arun Gupta PhD, MRSC Research Fellow Challis & Lewandowski Group Chemistry Department University of Warwick Gibbet Hill Road Coventry CV4 7AL United Kingdom

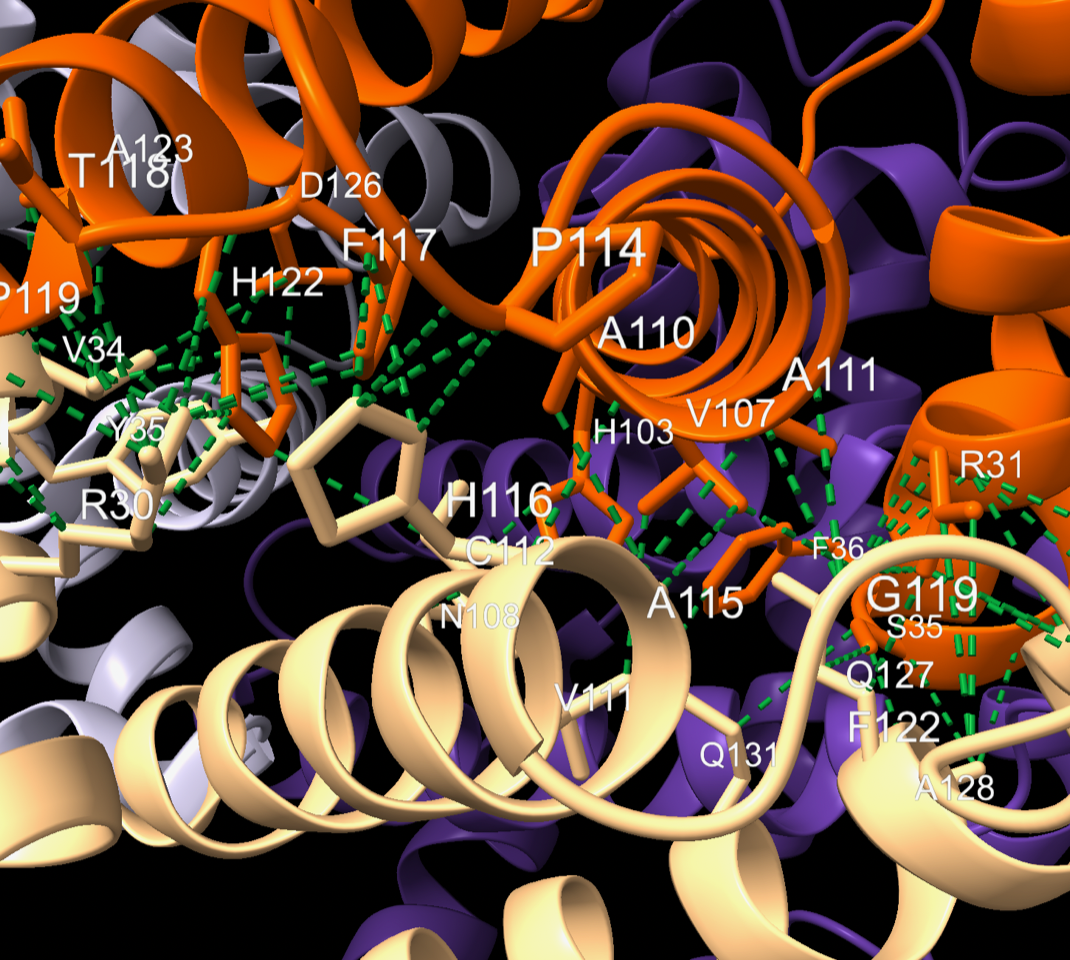
Hi Arun, I don't know what you mean by "check_interface" since that's not actually a ChimeraX command, but you did say custom, so maybe it's something you aliased. Anyway, assuming you have some residues of interest selected, your label command works just fine for me. So I'm also not sure what you mean by "not working"! Make sure you're actually using ChimeraX, of course. Test case, commands: open 4hhb hide rainbow chain palette puor contacts (/A & protein) restrict (/B & protein) sel true reveal true - the command above resulted in several selected residues. I showed these residues' sidechains and then used your label command, then cleared the selection: label sel text "{0.label_one_letter_code}{0.number}" ~select There are plenty of labeled residues, see screenshot of graphics window contents: Also, labeling isn't the only way to tell which residues were selected. The "contacts" command has an option to save all the pairwise atomic contact information to the Log and/or to a file. Or if you are using the "interfaces" (buried area) command to identify contacts, you can use it to create a selection and then a separate command "info" to write out a list of the selected atoms or residues. See the help for all of these commands: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/clashes.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/interfaces.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/info.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 9, 2024, at 6:49 AM, Gupta, Arun via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello ChimeraX team, I came to know this fabulous ChimeraX custom “check_interface” command to instantly identify interface residues between chains A and B of protein complex model #1. It works nicely with any number of protein complex models for e.g. #1, 2, 3, 4 with chains A and B However, when I am trying to “label” the identified interface residues using the below mentioned ChimeraX command for the various protein complex models #1, 2, 3, 4. It doesn’t seem to work. I am interested to identify and label the interface residues for the related set of various protein complex models #1, 2, 3,4. label sel text "{0.label_one_letter_code}{0.number}" Can somebody suggest what I am doing wrong with the ChimeraX “label” command ?? Thanks a lot ! Arun
{kind=link}
participants (2)
-
 Elaine Meng
Elaine Meng -
 Gupta, Arun
Gupta, Arun