how to vizualise hydrogen bonds between ligand and back-bone of protein?

Dear ChimeraX users! I am using the following command to calculate and save in the log information regarding hydrogen bonds based on the consideration of multi-frame pdb of the complex # calculate hydrogen bonds for the first 14 frames of pdb hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt which gives me something like this: structure_name: 18 H-bonds H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 ----------------------------------------------------------------------------- I noticed that in the log there is always information regarding interactions between the ligand and the side chains of the protein but nothing regarding hydrogen bonds involved backbone of the protein (Which is confirmed by X-ray data for my complex). for the test I used py@ol to visualise h-bonds and may see in the 12th frame the hydrogen bond involved backbone of the protein.. How could I modify my script to consider additional hydrogen bonds involving backbone atoms ? Thank you very much in advance! Cheers Enrico

Hello, Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear ChimeraX users! I am using the following command to calculate and save in the log information regarding hydrogen bonds based on the consideration of multi-frame pdb of the complex
# calculate hydrogen bonds for the first 14 frames of pdb hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
which gives me something like this: structure_name: 18 H-bonds H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 -----------------------------------------------------------------------------
I noticed that in the log there is always information regarding interactions between the ligand and the side chains of the protein but nothing regarding hydrogen bonds involved backbone of the protein (Which is confirmed by X-ray data for my complex). for the test I used py@ol to visualise h-bonds and may see in the 12th frame the hydrogen bond involved backbone of the protein.. How could I modify my script to consider additional hydrogen bonds involving backbone atoms ? Thank you very much in advance! Cheers Enrico
Thank you very much, Elaine! Indeed, I found that these two options influence the results. Cheers, Enrico чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>:
Hello, Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear ChimeraX users! I am using the following command to calculate and save in the log information regarding hydrogen bonds based on the consideration of multi-frame pdb of the complex
# calculate hydrogen bonds for the first 14 frames of pdb hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
which gives me something like this: structure_name: 18 H-bonds H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 -----------------------------------------------------------------------------
I noticed that in the log there is always information regarding interactions between the ligand and the side chains of the protein but nothing regarding hydrogen bonds involved backbone of the protein (Which is confirmed by X-ray data for my complex). for the test I used py@ol to visualise h-bonds and may see in the 12th frame the hydrogen bond involved backbone of the protein.. How could I modify my script to consider additional hydrogen bonds involving backbone atoms ? Thank you very much in advance! Cheers Enrico
Hello again! I've just make a test for several docking poses and I confirm that the hydrogen bond between ligand and backbone atoms never could be detected by chimera using hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt I tried to increase significantly the distance scope to 0.8 and the angle scope to 40 but the interactions could not be detected. Could it be the problem with the command that I am using ? Many thanks in advance! пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>:
Thank you very much, Elaine! Indeed, I found that these two options influence the results. Cheers, Enrico
чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>:
Hello, Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear ChimeraX users! I am using the following command to calculate and save in the log information regarding hydrogen bonds based on the consideration of multi-frame pdb of the complex
# calculate hydrogen bonds for the first 14 frames of pdb hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
which gives me something like this: structure_name: 18 H-bonds H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 -----------------------------------------------------------------------------
I noticed that in the log there is always information regarding interactions between the ligand and the side chains of the protein but nothing regarding hydrogen bonds involved backbone of the protein (Which is confirmed by X-ray data for my complex). for the test I used py@ol to visualise h-bonds and may see in the 12th frame the hydrogen bond involved backbone of the protein.. How could I modify my script to consider additional hydrogen bonds involving backbone atoms ? Thank you very much in advance! Cheers Enrico
Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8). Reasons to not find the H-bond: - maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem. - maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom. - maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it. However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello again! I've just make a test for several docking poses and I confirm that the hydrogen bond between ligand and backbone atoms never could be detected by chimera using hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
I tried to increase significantly the distance scope to 0.8 and the angle scope to 40 but the interactions could not be detected. Could it be the problem with the command that I am using ? Many thanks in advance!
пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>:
Thank you very much, Elaine! Indeed, I found that these two options influence the results. Cheers, Enrico
чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>:
Hello, Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear ChimeraX users! I am using the following command to calculate and save in the log information regarding hydrogen bonds based on the consideration of multi-frame pdb of the complex
# calculate hydrogen bonds for the first 14 frames of pdb hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
which gives me something like this: structure_name: 18 H-bonds H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 -----------------------------------------------------------------------------
I noticed that in the log there is always information regarding interactions between the ligand and the side chains of the protein but nothing regarding hydrogen bonds involved backbone of the protein (Which is confirmed by X-ray data for my complex). for the test I used py@ol to visualise h-bonds and may see in the 12th frame the hydrogen bond involved backbone of the protein.. How could I modify my script to consider additional hydrogen bonds involving backbone atoms ? Thank you very much in advance! Cheers Enrico
I see you are not displaying the H-bonds in your command anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed. In other words, if you are using the display to judge whether the H-bond is found you would need to use the "hbond" command options: makePseudobond true reveal true <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> Elaine
On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8).
Reasons to not find the H-bond:
- maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem.
- maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom.
- maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it.
However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond.
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello again! I've just make a test for several docking poses and I confirm that the hydrogen bond between ligand and backbone atoms never could be detected by chimera using hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
I tried to increase significantly the distance scope to 0.8 and the angle scope to 40 but the interactions could not be detected. Could it be the problem with the command that I am using ? Many thanks in advance!
пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>:
Thank you very much, Elaine! Indeed, I found that these two options influence the results. Cheers, Enrico
чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>:
Hello, Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear ChimeraX users! I am using the following command to calculate and save in the log information regarding hydrogen bonds based on the consideration of multi-frame pdb of the complex
# calculate hydrogen bonds for the first 14 frames of pdb hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
which gives me something like this: structure_name: 18 H-bonds H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 -----------------------------------------------------------------------------
I noticed that in the log there is always information regarding interactions between the ligand and the side chains of the protein but nothing regarding hydrogen bonds involved backbone of the protein (Which is confirmed by X-ray data for my complex). for the test I used py@ol to visualise h-bonds and may see in the 12th frame the hydrogen bond involved backbone of the protein.. How could I modify my script to consider additional hydrogen bonds involving backbone atoms ? Thank you very much in advance! Cheers Enrico
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users

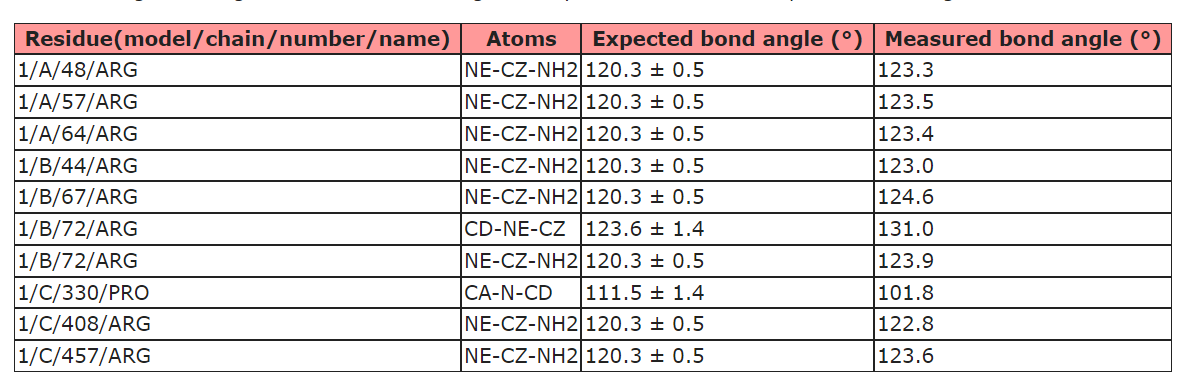
It seems that the parameterization of ISOLDE's force field differs from the metrics used by the official PBD validation server as I usually get complaints about Arg bond angles from the validation server (see below) even though everything seems fine in ISOLDE. Is there anything that I can/have to do about that? Or is that something that you would keep as it is and discuss with the PDB people? - Guido
{kind=link}

Hi Guido, Yes, there are a few small discrepancies between the AMBER force field and the libraries used for restraints/validation in refinement packages and the wwPDB. That's one of the reasons I try not to describe ISOLDE as a true refinement package (the other being that it currently doesn't perform B-factor refinement). You'll still want to run a final refinement with your package of choice before depositing to the wwPDB. To help with that, there are the commands: isolde write phenixRefineInput isolde write phenixRsrInput isolde write refmacRestraints Just type "usage isolde write" on the command line for more details on their use. Best, Tristan ________________________________ From: ChimeraX-users <chimerax-users-bounces@cgl.ucsf.edu> on behalf of Guido Hansen via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Sent: 16 March 2022 17:38 To: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: [chimerax-users] Bond angles of Arg in ISOLDE It seems that the parameterization of ISOLDE's force field differs from the metrics used by the official PBD validation server as I usually get complaints about Arg bond angles from the validation server (see below) even though everything seems fine in ISOLDE. Is there anything that I can/have to do about that? Or is that something that you would keep as it is and discuss with the PDB people? [cid:11d377ce14f2ac4dcd683be5424ebf5d@biochem.uni-luebeck.de] - Guido
{kind=link}
Hello Elaine Many thanks for these kind suggestions! In fact this back-bone H-bond has been validated by X-ray data (observed in several structures of my protein) and then by my docking studies. Please find enclosed the picture of this interaction: as we may see it is located on the relatively short distance between the O atom of the ligand and the HN of the backbone of the Glu residue. I believe it has to match both default slope criteria.... Regarding my command, actually I used it with batch version of chimeraX (in the script) so the goal was not to display the interactions but rather to save it directly into the log file: # calculate h-bonds between first 10 models from multi-model pdb file hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log_hbondsALL.log It produces a log with the correct H-bonds with the exemption of the interaction shown on the screenshot (which is always detected by other programs...) With best regards, Enrico вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>:
I see you are not displaying the H-bonds in your command anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed.
In other words, if you are using the display to judge whether the H-bond is found you would need to use the "hbond" command options:
makePseudobond true reveal true
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
Elaine
On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8).
Reasons to not find the H-bond:
- maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem.
- maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom.
- maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it.
However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond.
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello again! I've just make a test for several docking poses and I confirm that the hydrogen bond between ligand and backbone atoms never could be detected by chimera using hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
I tried to increase significantly the distance scope to 0.8 and the angle scope to 40 but the interactions could not be detected. Could it be the problem with the command that I am using ? Many thanks in advance!
пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>:
Thank you very much, Elaine! Indeed, I found that these two options influence the results. Cheers, Enrico
чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>:
Hello, Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear ChimeraX users! I am using the following command to calculate and save in the log information regarding hydrogen bonds based on the consideration of multi-frame pdb of the complex
# calculate hydrogen bonds for the first 14 frames of pdb hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
which gives me something like this: structure_name: 18 H-bonds H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 -----------------------------------------------------------------------------
I noticed that in the log there is always information regarding interactions between the ligand and the side chains of the protein but nothing regarding hydrogen bonds involved backbone of the protein (Which is confirmed by X-ray data for my complex). for the test I used py@ol to visualise h-bonds and may see in the 12th frame the hydrogen bond involved backbone of the protein.. How could I modify my script to consider additional hydrogen bonds involving backbone atoms ? Thank you very much in advance! Cheers Enrico
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
Hello, We can't figure out the reason for the "missing" H-bond unless you send us the file with the ligand and receptor atomic coordinates, the same ones shown in the image.. If you didn't want to share it with the whole chimerax-users list but you are willing to share it with the ChimeraX team, you can send it to just my individual e-mail address. However, if you need to keep it completely private we understand. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 21, 2022, at 2:28 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
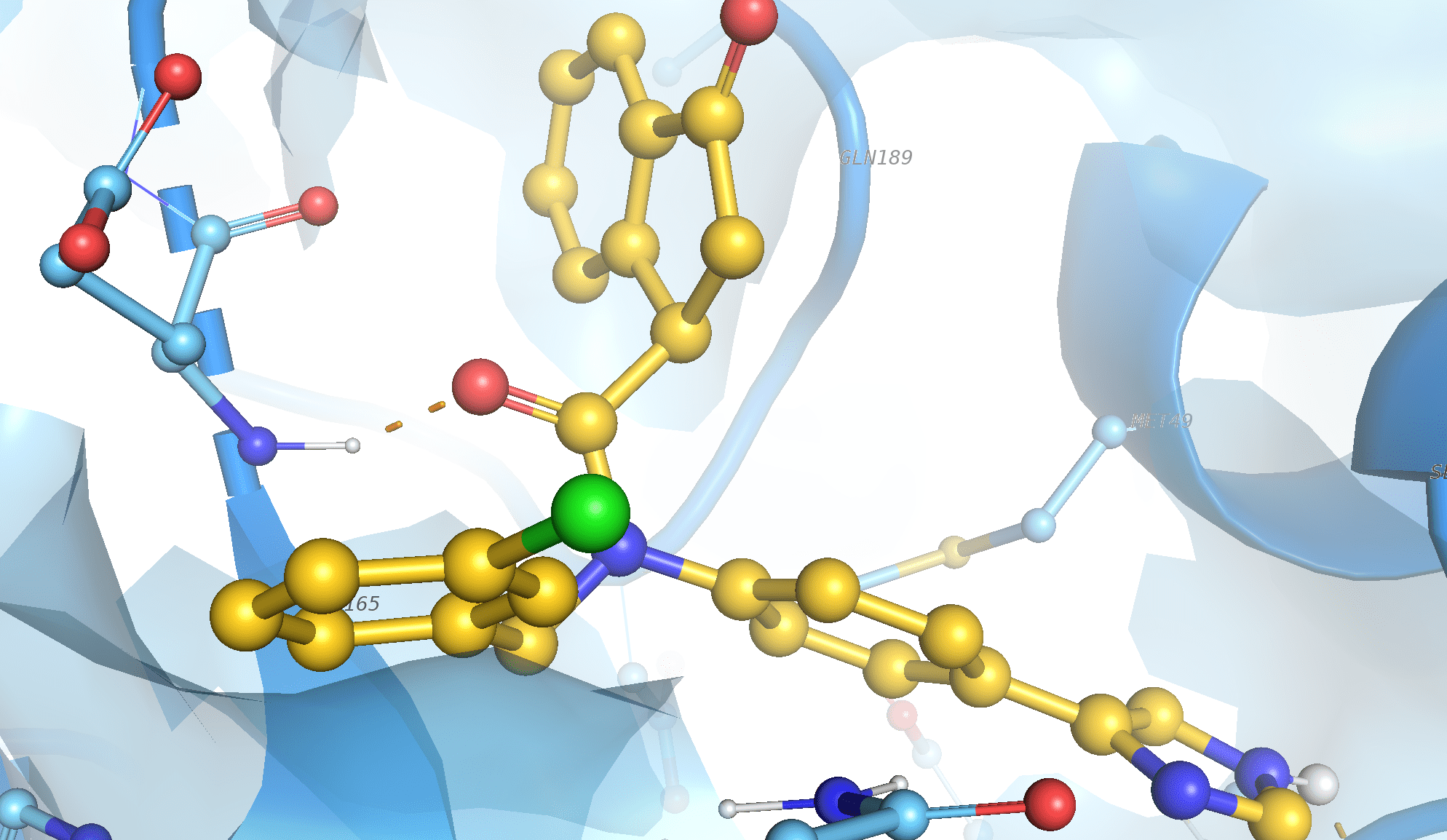
Hello Elaine Many thanks for these kind suggestions! In fact this back-bone H-bond has been validated by X-ray data (observed in several structures of my protein) and then by my docking studies. Please find enclosed the picture of this interaction: as we may see it is located on the relatively short distance between the O atom of the ligand and the HN of the backbone of the Glu residue. I believe it has to match both default slope criteria....
Regarding my command, actually I used it with batch version of chimeraX (in the script) so the goal was not to display the interactions but rather to save it directly into the log file:
# calculate h-bonds between first 10 models from multi-model pdb file hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log_hbondsALL.log
It produces a log with the correct H-bonds with the exemption of the interaction shown on the screenshot (which is always detected by other programs...) With best regards, Enrico
вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>:
I see you are not displaying the H-bonds in your command anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed.
In other words, if you are using the display to judge whether the H-bond is found you would need to use the "hbond" command options:
makePseudobond true reveal true
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
Elaine
On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8).
Reasons to not find the H-bond:
- maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem.
- maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom.
- maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it.
However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond.
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello again! I've just make a test for several docking poses and I confirm that the hydrogen bond between ligand and backbone atoms never could be detected by chimera using hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
I tried to increase significantly the distance scope to 0.8 and the angle scope to 40 but the interactions could not be detected. Could it be the problem with the command that I am using ? Many thanks in advance!
пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>:
Thank you very much, Elaine! Indeed, I found that these two options influence the results. Cheers, Enrico
чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>:
Hello, Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: > > Dear ChimeraX users! > I am using the following command to calculate and save in the log > information regarding hydrogen bonds based on the consideration of > multi-frame pdb of the complex > > # calculate hydrogen bonds for the first 14 frames of pdb > hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false > interModel false makePseudobonds false log true intraRes false > saveFile log.txt > > which gives me something like this: > structure_name: > 18 H-bonds > H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): > #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 > #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 > #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 > #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 > #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 > #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 > #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 > #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 > #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 > #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 > #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 > #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 > #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A > #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 > #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 > #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 > #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 > #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 > ----------------------------------------------------------------------------- > > I noticed that in the log there is always information regarding > interactions between the ligand and the side chains of the protein but > nothing regarding hydrogen bonds involved backbone of the protein > (Which is confirmed by X-ray data for my complex). for the test I used > py@ol to visualise h-bonds and may see in the 12th frame the hydrogen > bond involved backbone of the protein.. How could I modify my script > to consider additional hydrogen bonds involving backbone atoms ? > Thank you very much in advance! > Cheers > Enrico
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
<test_hbonds-min.png>
The problem is that your PDB file is fragmented, i.e. you only have some of the receptor residues so the backbone chain is broken in several places. When I try to run "hbonds" on what you sent me, there is a warning message in the Log: The following atoms were skipped as donors/acceptors due to missing heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; /? ASN 142 N; /? GLN 189 N When (for example) Glu 166 atom N does not have a bond from residue 165 atom C (because residue 165 is not in your structure), the correct angle for H-bonding is undetermined, so the hbonds algorithm ignores it. If you had the whole protein in the file, it would probably not be missing all these backbone bonds, and then it could correctly detect the H-bonds. Other programs probably have simple distance checking that doesn't use other atoms to figure out the proper angle. Whenever you get an unexpected result from a script, it is useful to try it interactively in the GUI so that you can see if there are any warning messages. I hope this clarifies the situation, Elaine
On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, We can't figure out the reason for the "missing" H-bond unless you send us the file with the ligand and receptor atomic coordinates, the same ones shown in the image.. If you didn't want to share it with the whole chimerax-users list but you are willing to share it with the ChimeraX team, you can send it to just my individual e-mail address. However, if you need to keep it completely private we understand. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 21, 2022, at 2:28 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello Elaine Many thanks for these kind suggestions! In fact this back-bone H-bond has been validated by X-ray data (observed in several structures of my protein) and then by my docking studies. Please find enclosed the picture of this interaction: as we may see it is located on the relatively short distance between the O atom of the ligand and the HN of the backbone of the Glu residue. I believe it has to match both default slope criteria....
Regarding my command, actually I used it with batch version of chimeraX (in the script) so the goal was not to display the interactions but rather to save it directly into the log file:
# calculate h-bonds between first 10 models from multi-model pdb file hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log_hbondsALL.log
It produces a log with the correct H-bonds with the exemption of the interaction shown on the screenshot (which is always detected by other programs...) With best regards, Enrico
вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>:
I see you are not displaying the H-bonds in your command anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed.
In other words, if you are using the display to judge whether the H-bond is found you would need to use the "hbond" command options:
makePseudobond true reveal true
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
Elaine
On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8).
Reasons to not find the H-bond:
- maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem.
- maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom.
- maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it.
However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond.
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello again! I've just make a test for several docking poses and I confirm that the hydrogen bond between ligand and backbone atoms never could be detected by chimera using hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
I tried to increase significantly the distance scope to 0.8 and the angle scope to 40 but the interactions could not be detected. Could it be the problem with the command that I am using ? Many thanks in advance!
пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>:
Thank you very much, Elaine! Indeed, I found that these two options influence the results. Cheers, Enrico
чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: > > Hello, > Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein. > > <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> > > Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive. > > <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> > > I hope this helps, > Elaine > ----- > Elaine C. Meng, Ph.D. > UCSF Chimera(X) team > Department of Pharmaceutical Chemistry > University of California, San Francisco > >> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >> >> Dear ChimeraX users! >> I am using the following command to calculate and save in the log >> information regarding hydrogen bonds based on the consideration of >> multi-frame pdb of the complex >> >> # calculate hydrogen bonds for the first 14 frames of pdb >> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >> interModel false makePseudobonds false log true intraRes false >> saveFile log.txt >> >> which gives me something like this: >> structure_name: >> 18 H-bonds >> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 >> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 >> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 >> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 >> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 >> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 >> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 >> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 >> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 >> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 >> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 >> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 >> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A >> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 >> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 >> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 >> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 >> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 >> ----------------------------------------------------------------------------- >> >> I noticed that in the log there is always information regarding >> interactions between the ligand and the side chains of the protein but >> nothing regarding hydrogen bonds involved backbone of the protein >> (Which is confirmed by X-ray data for my complex). for the test I used >> py@ol to visualise h-bonds and may see in the 12th frame the hydrogen >> bond involved backbone of the protein.. How could I modify my script >> to consider additional hydrogen bonds involving backbone atoms ? >> Thank you very much in advance! >> Cheers >> Enrico >
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
<test_hbonds-min.png>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Right, thank you very much Elaine! Now I gotcha! Indeed it should be the case since the pdb was produced by the concatenation of the autodock results, (which left flexible residues with the ligand) with the receptor pdb w/o these side-chains. BTW I double checked and could reproduce the hydrogen bond with the side-chain atoms of the same residue, so the problem indeed is related only to backbone (particularly to the N atom).. Anyway thank you very much for the help! Cheers, Enrico пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>:
The problem is that your PDB file is fragmented, i.e. you only have some of the receptor residues so the backbone chain is broken in several places. When I try to run "hbonds" on what you sent me, there is a warning message in the Log:
The following atoms were skipped as donors/acceptors due to missing heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; /? ASN 142 N; /? GLN 189 N
When (for example) Glu 166 atom N does not have a bond from residue 165 atom C (because residue 165 is not in your structure), the correct angle for H-bonding is undetermined, so the hbonds algorithm ignores it. If you had the whole protein in the file, it would probably not be missing all these backbone bonds, and then it could correctly detect the H-bonds.
Other programs probably have simple distance checking that doesn't use other atoms to figure out the proper angle.
Whenever you get an unexpected result from a script, it is useful to try it interactively in the GUI so that you can see if there are any warning messages.
I hope this clarifies the situation, Elaine
On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, We can't figure out the reason for the "missing" H-bond unless you send us the file with the ligand and receptor atomic coordinates, the same ones shown in the image.. If you didn't want to share it with the whole chimerax-users list but you are willing to share it with the ChimeraX team, you can send it to just my individual e-mail address. However, if you need to keep it completely private we understand. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 21, 2022, at 2:28 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello Elaine Many thanks for these kind suggestions! In fact this back-bone H-bond has been validated by X-ray data (observed in several structures of my protein) and then by my docking studies. Please find enclosed the picture of this interaction: as we may see it is located on the relatively short distance between the O atom of the ligand and the HN of the backbone of the Glu residue. I believe it has to match both default slope criteria....
Regarding my command, actually I used it with batch version of chimeraX (in the script) so the goal was not to display the interactions but rather to save it directly into the log file:
# calculate h-bonds between first 10 models from multi-model pdb file hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log_hbondsALL.log
It produces a log with the correct H-bonds with the exemption of the interaction shown on the screenshot (which is always detected by other programs...) With best regards, Enrico
вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>:
I see you are not displaying the H-bonds in your command anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed.
In other words, if you are using the display to judge whether the H-bond is found you would need to use the "hbond" command options:
makePseudobond true reveal true
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
Elaine
On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8).
Reasons to not find the H-bond:
- maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem.
- maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom.
- maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it.
However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond.
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello again! I've just make a test for several docking poses and I confirm that the hydrogen bond between ligand and backbone atoms never could be detected by chimera using hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log.txt
I tried to increase significantly the distance scope to 0.8 and the angle scope to 40 but the interactions could not be detected. Could it be the problem with the command that I am using ? Many thanks in advance!
пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>: > > Thank you very much, Elaine! > Indeed, I found that these two options influence the results. > Cheers, > Enrico > > чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: >> >> Hello, >> Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein. >> >> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> >> >> Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive. >> >> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> >> >> I hope this helps, >> Elaine >> ----- >> Elaine C. Meng, Ph.D. >> UCSF Chimera(X) team >> Department of Pharmaceutical Chemistry >> University of California, San Francisco >> >>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >>> >>> Dear ChimeraX users! >>> I am using the following command to calculate and save in the log >>> information regarding hydrogen bonds based on the consideration of >>> multi-frame pdb of the complex >>> >>> # calculate hydrogen bonds for the first 14 frames of pdb >>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>> interModel false makePseudobonds false log true intraRes false >>> saveFile log.txt >>> >>> which gives me something like this: >>> structure_name: >>> 18 H-bonds >>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 >>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 >>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 >>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 >>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 >>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 >>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 >>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 >>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 >>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 >>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 >>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 >>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A >>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 >>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 >>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 >>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 >>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 >>> ----------------------------------------------------------------------------- >>> >>> I noticed that in the log there is always information regarding >>> interactions between the ligand and the side chains of the protein but >>> nothing regarding hydrogen bonds involved backbone of the protein >>> (Which is confirmed by X-ray data for my complex). for the test I used >>> py@ol to visualise h-bonds and may see in the 12th frame the hydrogen >>> bond involved backbone of the protein.. How could I modify my script >>> to consider additional hydrogen bonds involving backbone atoms ? >>> Thank you very much in advance! >>> Cheers >>> Enrico >>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
<test_hbonds-min.png>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users

Hi Enrico, You may want to try rebuilding these parts with modeller or some other template-based modelling method. There may also be an option to refine the docked structure in autodock like there is in HADDOCK or an ability to use HADDOCK for the autodock output. Best wishes James Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> escribió:
Right, thank you very much Elaine! Now I gotcha! Indeed it should be the case since the pdb was produced by the concatenation of the autodock results, (which left flexible residues with the ligand) with the receptor pdb w/o these side-chains. BTW I double checked and could reproduce the hydrogen bond with the side-chain atoms of the same residue, so the problem indeed is related only to backbone (particularly to the N atom).. Anyway thank you very much for the help! Cheers, Enrico
пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>:
The problem is that your PDB file is fragmented, i.e. you only have some of the receptor residues so the backbone chain is broken in several places. When I try to run "hbonds" on what you sent me, there is a warning message in the Log:
The following atoms were skipped as donors/acceptors due to missing heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; /? ASN 142 N; /? GLN 189 N
When (for example) Glu 166 atom N does not have a bond from residue 165 atom C (because residue 165 is not in your structure), the correct angle for H-bonding is undetermined, so the hbonds algorithm ignores it. If you had the whole protein in the file, it would probably not be missing all these backbone bonds, and then it could correctly detect the H-bonds.
Other programs probably have simple distance checking that doesn't use other atoms to figure out the proper angle.
Whenever you get an unexpected result from a script, it is useful to try it interactively in the GUI so that you can see if there are any warning messages.
I hope this clarifies the situation, Elaine
On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, We can't figure out the reason for the "missing" H-bond unless you send us the file with the ligand and receptor atomic coordinates, the same ones shown in the image.. If you didn't want to share it with the whole chimerax-users list but you are willing to share it with the ChimeraX team, you can send it to just my individual e-mail address. However, if you need to keep it completely private we understand. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 21, 2022, at 2:28 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello Elaine Many thanks for these kind suggestions! In fact this back-bone H-bond has been validated by X-ray data (observed in several structures of my protein) and then by my docking studies. Please find enclosed the picture of this interaction: as we may see it is located on the relatively short distance between the O atom of the ligand and the HN of the backbone of the Glu residue. I believe it has to match both default slope criteria....
Regarding my command, actually I used it with batch version of chimeraX (in the script) so the goal was not to display the interactions but rather to save it directly into the log file:
# calculate h-bonds between first 10 models from multi-model pdb file hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log_hbondsALL.log
It produces a log with the correct H-bonds with the exemption of the interaction shown on the screenshot (which is always detected by other programs...) With best regards, Enrico
вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>:
I see you are not displaying the H-bonds in your command
anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed.
In other words, if you are using the display to judge whether
the H-bond is found you would need to use the "hbond" command options:
makePseudobond true reveal true
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
Elaine
On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users
<chimerax-users@cgl.ucsf.edu> wrote:
Those "slop" values are tolerances, not the scope (cutoff):
i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8).
Reasons to not find the H-bond:
- maybe your atom specification is wrong... in fact I have no
idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem.
- maybe those types of atoms are not considered by the H-bond
detection. I.e. it does not consider C to be an H-bonding type of atom.
- maybe it is because the H-bond geometry is very poor, you
still need to increase the slop values even more to find it.
However: We believe that Chimera and ChimeraX provide
high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond.
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
> On Mar 15, 2022, at 8:17 AM, Enrico Martinez
<jmsstarlight@gmail.com> wrote:
> > Hello again! > I've just make a test for several docking poses and I confirm that the > hydrogen bond between ligand and backbone atoms never could be > detected by chimera using > hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false > interModel false makePseudobonds false log true intraRes false > saveFile log.txt > > I tried to increase significantly the distance scope to 0.8 and the > angle scope to 40 but the interactions could not be detected. Could it > be the problem with the command that I am using ? > Many thanks in advance! > > пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>: >> >> Thank you very much, Elaine! >> Indeed, I found that these two options influence the results. >> Cheers, >> Enrico >> >> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: >>> >>> Hello, >>> Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein. >>> >>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> >>> >>> Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive. >>> >>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> >>> >>> I hope this helps, >>> Elaine >>> ----- >>> Elaine C. Meng, Ph.D. >>> UCSF Chimera(X) team >>> Department of Pharmaceutical Chemistry >>> University of California, San Francisco >>> >>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >>>> >>>> Dear ChimeraX users! >>>> I am using the following command to calculate and save in the log >>>> information regarding hydrogen bonds based on the consideration of >>>> multi-frame pdb of the complex >>>> >>>> # calculate hydrogen bonds for the first 14 frames of pdb >>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>>> interModel false makePseudobonds false log true intraRes false >>>> saveFile log.txt >>>> >>>> which gives me something like this: >>>> structure_name: >>>> 18 H-bonds >>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 >>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 >>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 >>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 >>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 >>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 >>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 >>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 >>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 >>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 >>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 >>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 >>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A >>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 >>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 >>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 >>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 >>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 >>>>
>>>> >>>> I noticed that in the log there is always information regarding >>>> interactions between the ligand and the side chains of the protein but >>>> nothing regarding hydrogen bonds involved backbone of the protein >>>> (Which is confirmed by X-ray data for my complex). for the test I used >>>> py@ol to visualise h-bonds and may see in the 12th frame the hydrogen >>>> bond involved backbone of the protein.. How could I modify my script >>>> to consider additional hydrogen bonds involving backbone atoms ? >>>> Thank you very much in advance! >>>> Cheers >>>> Enrico >>> >
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
<test_hbonds-min.png>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi James, many thanks for these suggestions! Indeed I am looking for some utility that may be useful to fix my "fragmented" multi-model pdb, converting it into "common" format (considering all frames). I've just made a quick test excluding the residues where I had difficulties with the visualisation of the "back-bone" hydrogen bond from the list of the "flexible residues" in VINA calculations and here it is: since this part is no more "fragmented", Chimera is able to find the hydrogen bond with the default "slopes" values. Victory! May be there is some command in Chimera that may fix the problem? Essentially I am dealing with protein-ligand complex produced by the CAT of two initial pdbs: i) multi-model pdb of the docking poses (containing ligand and 4-5 side-chains) + the "static part" of the protein (all of the atoms excluding these 4-5 flexible side-chains) merged into the each model. It looks absolutely correctly while examining it in any molecular vizualisator so the problem could only be detected in ChimeraX.. I would be grateful for any suggestions Cheers, Enrico вт, 22 мар. 2022 г. в 12:54, JAMES MICHAEL S1JJRUdFUiA= via ChimeraX-users <chimerax-users@cgl.ucsf.edu>:
Hi Enrico, You may want to try rebuilding these parts with modeller or some other template-based modelling method. There may also be an option to refine the docked structure in autodock like there is in HADDOCK or an ability to use HADDOCK for the autodock output. Best wishes James
Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> escribió:
Right, thank you very much Elaine! Now I gotcha! Indeed it should be the case since the pdb was produced by the concatenation of the autodock results, (which left flexible residues with the ligand) with the receptor pdb w/o these side-chains. BTW I double checked and could reproduce the hydrogen bond with the side-chain atoms of the same residue, so the problem indeed is related only to backbone (particularly to the N atom).. Anyway thank you very much for the help! Cheers, Enrico
пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>:
The problem is that your PDB file is fragmented, i.e. you only have some of the receptor residues so the backbone chain is broken in several places. When I try to run "hbonds" on what you sent me, there is a warning message in the Log:
The following atoms were skipped as donors/acceptors due to missing heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; /? ASN 142 N; /? GLN 189 N
When (for example) Glu 166 atom N does not have a bond from residue 165 atom C (because residue 165 is not in your structure), the correct angle for H-bonding is undetermined, so the hbonds algorithm ignores it. If you had the whole protein in the file, it would probably not be missing all these backbone bonds, and then it could correctly detect the H-bonds.
Other programs probably have simple distance checking that doesn't use other atoms to figure out the proper angle.
Whenever you get an unexpected result from a script, it is useful to try it interactively in the GUI so that you can see if there are any warning messages.
I hope this clarifies the situation, Elaine
On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, We can't figure out the reason for the "missing" H-bond unless you send us the file with the ligand and receptor atomic coordinates, the same ones shown in the image.. If you didn't want to share it with the whole chimerax-users list but you are willing to share it with the ChimeraX team, you can send it to just my individual e-mail address. However, if you need to keep it completely private we understand. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 21, 2022, at 2:28 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello Elaine Many thanks for these kind suggestions! In fact this back-bone H-bond has been validated by X-ray data (observed in several structures of my protein) and then by my docking studies. Please find enclosed the picture of this interaction: as we may see it is located on the relatively short distance between the O atom of the ligand and the HN of the backbone of the Glu residue. I believe it has to match both default slope criteria....
Regarding my command, actually I used it with batch version of chimeraX (in the script) so the goal was not to display the interactions but rather to save it directly into the log file:
# calculate h-bonds between first 10 models from multi-model pdb file hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log_hbondsALL.log
It produces a log with the correct H-bonds with the exemption of the interaction shown on the screenshot (which is always detected by other programs...) With best regards, Enrico
вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>:
I see you are not displaying the H-bonds in your command
anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed.
In other words, if you are using the display to judge whether
the H-bond is found you would need to use the "hbond" command options:
makePseudobond true reveal true
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
Elaine
> On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users
<chimerax-users@cgl.ucsf.edu> wrote:
> > Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8). > > Reasons to not find the H-bond: > > - maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem. > > - maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom. > > - maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it. > > However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond. > > Best, > Elaine > ----- > Elaine C. Meng, Ph.D. > UCSF Chimera(X) team > Department of Pharmaceutical Chemistry > University of California, San Francisco > >> On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote: >> >> Hello again! >> I've just make a test for several docking poses and I confirm that the >> hydrogen bond between ligand and backbone atoms never could be >> detected by chimera using >> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >> interModel false makePseudobonds false log true intraRes false >> saveFile log.txt >> >> I tried to increase significantly the distance scope to 0.8 and the >> angle scope to 40 but the interactions could not be detected. Could it >> be the problem with the command that I am using ? >> Many thanks in advance! >> >> пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>: >>> >>> Thank you very much, Elaine! >>> Indeed, I found that these two options influence the results. >>> Cheers, >>> Enrico >>> >>> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: >>>> >>>> Hello, >>>> Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein. >>>> >>>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> >>>> >>>> Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive. >>>> >>>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> >>>> >>>> I hope this helps, >>>> Elaine >>>> ----- >>>> Elaine C. Meng, Ph.D. >>>> UCSF Chimera(X) team >>>> Department of Pharmaceutical Chemistry >>>> University of California, San Francisco >>>> >>>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >>>>> >>>>> Dear ChimeraX users! >>>>> I am using the following command to calculate and save in the log >>>>> information regarding hydrogen bonds based on the consideration of >>>>> multi-frame pdb of the complex >>>>> >>>>> # calculate hydrogen bonds for the first 14 frames of pdb >>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>>>> interModel false makePseudobonds false log true intraRes false >>>>> saveFile log.txt >>>>> >>>>> which gives me something like this: >>>>> structure_name: >>>>> 18 H-bonds >>>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >>>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 >>>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 >>>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 >>>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 >>>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 >>>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 >>>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 >>>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 >>>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 >>>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 >>>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 >>>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 >>>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A >>>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 >>>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 >>>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 >>>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 >>>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 >>>>>
>>>>> >>>>> I noticed that in the log there is always information regarding >>>>> interactions between the ligand and the side chains of the protein but >>>>> nothing regarding hydrogen bonds involved backbone of the protein >>>>> (Which is confirmed by X-ray data for my complex). for the test I used >>>>> py@ol to visualise h-bonds and may see in the 12th frame the hydrogen >>>>> bond involved backbone of the protein.. How could I modify my script >>>>> to consider additional hydrogen bonds involving backbone atoms ? >>>>> Thank you very much in advance! >>>>> Cheers >>>>> Enrico >>>> >> > > > _______________________________________________ > ChimeraX-users mailing list > ChimeraX-users@cgl.ucsf.edu > Manage subscription: > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >
<test_hbonds-min.png>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi Enrico, It would probably be better to combine them model by model rather than just cat-ing the two. Probably the ProDy Python API could handle this but I'm not exactly sure. You could try something along the lines of the following. I just made some PDB files that sound like what you described and it worked fine. from prody import * atoms1 = parsePDB(pdbfile1) # multi-model will be read as multiple coordinate sets atoms2 = parsePDB(pdbfile2) # I'm assuming it just has one coordinate set atoms3 = atoms2.copy() for i in range(atoms1.numCoordsets()-1): atoms3.addCoordset(atoms2) atoms4 = atoms1 + atoms3 writePDB(pdbfile3, atoms4) You'd also need to open the resulting PDB file in pymol and save it again (making sure to include all states) to get the atoms back in the right order for chimerax to know they belong to the save residues. Best wishes James Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hi James, many thanks for these suggestions! Indeed I am looking for some utility that may be useful to fix my "fragmented" multi-model pdb, converting it into "common" format (considering all frames). I've just made a quick test excluding the residues where I had difficulties with the visualisation of the "back-bone" hydrogen bond from the list of the "flexible residues" in VINA calculations and here it is: since this part is no more "fragmented", Chimera is able to find the hydrogen bond with the default "slopes" values. Victory! May be there is some command in Chimera that may fix the problem? Essentially I am dealing with protein-ligand complex produced by the CAT of two initial pdbs: i) multi-model pdb of the docking poses (containing ligand and 4-5 side-chains) + the "static part" of the protein (all of the atoms excluding these 4-5 flexible side-chains) merged into the each model. It looks absolutely correctly while examining it in any molecular vizualisator so the problem could only be detected in ChimeraX.. I would be grateful for any suggestions Cheers, Enrico
вт, 22 мар. 2022 г. в 12:54, JAMES MICHAEL S1JJRUdFUiA= via ChimeraX-users <chimerax-users@cgl.ucsf.edu>:
Hi Enrico, You may want to try rebuilding these parts with modeller or some other template-based modelling method. There may also be an option to refine the docked structure in autodock like there is in HADDOCK or an ability to use HADDOCK for the autodock output. Best wishes James
Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> escribió:
Right, thank you very much Elaine! Now I gotcha! Indeed it should be the case since the pdb was produced by the concatenation of the autodock results, (which left flexible residues with the ligand) with the receptor pdb w/o these side-chains. BTW I double checked and could reproduce the hydrogen bond with the side-chain atoms of the same residue, so the problem indeed is related only to backbone (particularly to the N atom).. Anyway thank you very much for the help! Cheers, Enrico
пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>:
The problem is that your PDB file is fragmented, i.e. you only have some of the receptor residues so the backbone chain is broken in several places. When I try to run "hbonds" on what you sent me, there is a warning message in the Log:
The following atoms were skipped as donors/acceptors due to missing heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; /? ASN 142 N; /? GLN 189 N
When (for example) Glu 166 atom N does not have a bond from residue 165 atom C (because residue 165 is not in your structure), the correct angle for H-bonding is undetermined, so the hbonds algorithm ignores it. If you had the whole protein in the file, it would probably not be missing all these backbone bonds, and then it could correctly detect the H-bonds.
Other programs probably have simple distance checking that doesn't use other atoms to figure out the proper angle.
Whenever you get an unexpected result from a script, it is useful to try it interactively in the GUI so that you can see if there are any warning messages.
I hope this clarifies the situation, Elaine
On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, We can't figure out the reason for the "missing" H-bond unless you send us the file with the ligand and receptor atomic coordinates, the same ones shown in the image.. If you didn't want to share it with the whole chimerax-users list but you are willing to share it with the ChimeraX team, you can send it to just my individual e-mail address. However, if you need to keep it completely private we understand. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 21, 2022, at 2:28 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote:
Hello Elaine Many thanks for these kind suggestions! In fact this back-bone H-bond has been validated by X-ray data (observed in several structures of my protein) and then by my docking studies. Please find enclosed the picture of this interaction: as we may see it is located on the relatively short distance between the O atom of the ligand and the HN of the backbone of the Glu residue. I believe it has to match both default slope criteria....
Regarding my command, actually I used it with batch version of chimeraX (in the script) so the goal was not to display the interactions but rather to save it directly into the log file:
# calculate h-bonds between first 10 models from multi-model pdb file hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false interModel false makePseudobonds false log true intraRes false saveFile log_hbondsALL.log
It produces a log with the correct H-bonds with the exemption of the interaction shown on the screenshot (which is always detected by other programs...) With best regards, Enrico
вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>: > > I see you are not displaying the H-bonds in your command anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed. > > In other words, if you are using the display to judge whether the H-bond is found you would need to use the "hbond" command options: > > makePseudobond true reveal true > >
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
> > Elaine > > >> On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >> >> Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8). >> >> Reasons to not find the H-bond: >> >> - maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem. >> >> - maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom. >> >> - maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it. >> >> However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond. >> >> Best, >> Elaine >> ----- >> Elaine C. Meng, Ph.D. >> UCSF Chimera(X) team >> Department of Pharmaceutical Chemistry >> University of California, San Francisco >> >>> On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote: >>> >>> Hello again! >>> I've just make a test for several docking poses and I confirm that the >>> hydrogen bond between ligand and backbone atoms never could be >>> detected by chimera using >>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>> interModel false makePseudobonds false log true intraRes false >>> saveFile log.txt >>> >>> I tried to increase significantly the distance scope to 0.8 and the >>> angle scope to 40 but the interactions could not be detected. Could it >>> be the problem with the command that I am using ? >>> Many thanks in advance! >>> >>> пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>: >>>> >>>> Thank you very much, Elaine! >>>> Indeed, I found that these two options influence the results. >>>> Cheers, >>>> Enrico >>>> >>>> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: >>>>> >>>>> Hello, >>>>> Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein. >>>>> >>>>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> >>>>> >>>>> Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive. >>>>> >>>>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> >>>>> >>>>> I hope this helps, >>>>> Elaine >>>>> ----- >>>>> Elaine C. Meng, Ph.D. >>>>> UCSF Chimera(X) team >>>>> Department of Pharmaceutical Chemistry >>>>> University of California, San Francisco >>>>> >>>>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >>>>>> >>>>>> Dear ChimeraX users! >>>>>> I am using the following command to calculate and save in the log >>>>>> information regarding hydrogen bonds based on the consideration of >>>>>> multi-frame pdb of the complex >>>>>> >>>>>> # calculate hydrogen bonds for the first 14 frames of pdb >>>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>>>>> interModel false makePseudobonds false log true intraRes false >>>>>> saveFile log.txt >>>>>> >>>>>> which gives me something like this: >>>>>> structure_name: >>>>>> 18 H-bonds >>>>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >>>>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 >>>>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 >>>>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 >>>>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 >>>>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 >>>>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 >>>>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 >>>>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 >>>>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 >>>>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 >>>>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 >>>>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 >>>>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A >>>>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 >>>>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 >>>>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 >>>>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 >>>>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 >>>>>>
>>>>>> >>>>>> I noticed that in the log there is always information regarding >>>>>> interactions between the ligand and the side chains of the protein but >>>>>> nothing regarding hydrogen bonds involved backbone of the protein >>>>>> (Which is confirmed by X-ray data for my complex). for the test I used >>>>>> py@ol to visualise h-bonds and may see in the 12th frame the hydrogen >>>>>> bond involved backbone of the protein.. How could I modify my script >>>>>> to consider additional hydrogen bonds involving backbone atoms ? >>>>>> Thank you very much in advance! >>>>>> Cheers >>>>>> Enrico >>>>> >>> >> >> >> _______________________________________________ >> ChimeraX-users mailing list >> ChimeraX-users@cgl.ucsf.edu >> Manage subscription: >> https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >> > <test_hbonds-min.png>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hey James, actually, it seems to me that I have already done it: copy/past a receptor model (with the static residues!) into EACH of the model containing docking pose of the ligand + 5 flexible residues. Here is my script to produce multi-model pdb with the complex: awk 'NR==FNR {s = s $0 ORS; next} $0 == "ENDMDL" {$0 = s $0} 1' "${temp}"/static_receptor.pdb "${results}"/docking_poses_with_flexible_residues.pdb >> "${results}"/complex. pdb basically it looks for the ENDMDL term in each frame of the multi-model pdb and past there the model of the receptor. So the both are present in each frames. The issue is related to the Flexible residues, which are always near the ligand atoms (since they are present in the each model with the docking poses) so the resulted "combined" PDB appeared to be "fragmented" in the sence of the side-chains of the flexible residues (taken from the first pdb) as well as their backbone atoms (present in another pdb with the static receptor), so .. Although the combined PDB produced by my script seems to be absolutely normal, there are problems with the recognition of the back-bone atoms of the flexible residues, which are not considered as the donors for the hydrogen bonds ... may-be there is some program which may just open my pdb and convert it (the order of the string) into the normal format since the problem is always related to the position of the flexible side-chains in each frame... Enrico вт, 22 мар. 2022 г. в 16:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico, It would probably be better to combine them model by model rather than just cat-ing the two. Probably the ProDy Python API could handle this but I'm not exactly sure. You could try something along the lines of the following. I just made some PDB files that sound like what you described and it worked fine.
from prody import *
atoms1 = parsePDB(pdbfile1) # multi-model will be read as multiple coordinate sets
atoms2 = parsePDB(pdbfile2) # I'm assuming it just has one coordinate set
atoms3 = atoms2.copy()
for i in range(atoms1.numCoordsets()-1): atoms3.addCoordset(atoms2)
atoms4 = atoms1 + atoms3
writePDB(pdbfile3, atoms4)
You'd also need to open the resulting PDB file in pymol and save it again (making sure to include all states) to get the atoms back in the right order for chimerax to know they belong to the save residues.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hi James, many thanks for these suggestions! Indeed I am looking for some utility that may be useful to fix my "fragmented" multi-model pdb, converting it into "common" format (considering all frames). I've just made a quick test excluding the residues where I had difficulties with the visualisation of the "back-bone" hydrogen bond from the list of the "flexible residues" in VINA calculations and here it is: since this part is no more "fragmented", Chimera is able to find the hydrogen bond with the default "slopes" values. Victory! May be there is some command in Chimera that may fix the problem? Essentially I am dealing with protein-ligand complex produced by the CAT of two initial pdbs: i) multi-model pdb of the docking poses (containing ligand and 4-5 side-chains) + the "static part" of the protein (all of the atoms excluding these 4-5 flexible side-chains) merged into the each model. It looks absolutely correctly while examining it in any molecular vizualisator so the problem could only be detected in ChimeraX.. I would be grateful for any suggestions Cheers, Enrico
вт, 22 мар. 2022 г. в 12:54, JAMES MICHAEL S1JJRUdFUiA= via ChimeraX-users <chimerax-users@cgl.ucsf.edu>:
Hi Enrico, You may want to try rebuilding these parts with modeller or some other template-based modelling method. There may also be an option to refine the docked structure in autodock like there is in HADDOCK or an ability to use HADDOCK for the autodock output. Best wishes James
Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> escribió:
Right, thank you very much Elaine! Now I gotcha! Indeed it should be the case since the pdb was produced by the concatenation of the autodock results, (which left flexible residues with the ligand) with the receptor pdb w/o these side-chains. BTW I double checked and could reproduce the hydrogen bond with the side-chain atoms of the same residue, so the problem indeed is related only to backbone (particularly to the N atom).. Anyway thank you very much for the help! Cheers, Enrico
пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>:
The problem is that your PDB file is fragmented, i.e. you only have some of the receptor residues so the backbone chain is broken in several places. When I try to run "hbonds" on what you sent me, there is a warning message in the Log:
The following atoms were skipped as donors/acceptors due to missing heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; /? ASN 142 N; /? GLN 189 N
When (for example) Glu 166 atom N does not have a bond from residue 165 atom C (because residue 165 is not in your structure), the correct angle for H-bonding is undetermined, so the hbonds algorithm ignores it. If you had the whole protein in the file, it would probably not be missing all these backbone bonds, and then it could correctly detect the H-bonds.
Other programs probably have simple distance checking that doesn't use other atoms to figure out the proper angle.
Whenever you get an unexpected result from a script, it is useful to try it interactively in the GUI so that you can see if there are any warning messages.
I hope this clarifies the situation, Elaine
On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, We can't figure out the reason for the "missing" H-bond unless you send us the file with the ligand and receptor atomic coordinates, the same ones shown in the image.. If you didn't want to share it with the whole chimerax-users list but you are willing to share it with the ChimeraX team, you can send it to just my individual e-mail address. However, if you need to keep it completely private we understand. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
> On Mar 21, 2022, at 2:28 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote: > > Hello Elaine > Many thanks for these kind suggestions! > In fact this back-bone H-bond has been validated by X-ray data > (observed in several structures of my protein) and then by my docking > studies. Please find enclosed the picture of this interaction: as we > may see it is located on the relatively short distance between the O > atom of the ligand and the HN of the backbone of the Glu residue. I > believe it has to match both default slope criteria.... > > Regarding my command, actually I used it with batch version of > chimeraX (in the script) so the goal was not to display the > interactions but rather to save it directly into the log file: > > # calculate h-bonds between first 10 models from multi-model pdb file > hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false > interModel false makePseudobonds false log true intraRes false > saveFile log_hbondsALL.log > > It produces a log with the correct H-bonds with the exemption of the > interaction shown on the screenshot (which is always detected by other > programs...) > With best regards, > Enrico > > вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>: >> >> I see you are not displaying the H-bonds in your command anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed. >> >> In other words, if you are using the display to judge whether the H-bond is found you would need to use the "hbond" command options: >> >> makePseudobond true reveal true >> >>
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
>> >> Elaine >> >> >>> On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >>> >>> Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8). >>> >>> Reasons to not find the H-bond: >>> >>> - maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem. >>> >>> - maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom. >>> >>> - maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it. >>> >>> However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond. >>> >>> Best, >>> Elaine >>> ----- >>> Elaine C. Meng, Ph.D. >>> UCSF Chimera(X) team >>> Department of Pharmaceutical Chemistry >>> University of California, San Francisco >>> >>>> On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote: >>>> >>>> Hello again! >>>> I've just make a test for several docking poses and I confirm that the >>>> hydrogen bond between ligand and backbone atoms never could be >>>> detected by chimera using >>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>>> interModel false makePseudobonds false log true intraRes false >>>> saveFile log.txt >>>> >>>> I tried to increase significantly the distance scope to 0.8 and the >>>> angle scope to 40 but the interactions could not be detected. Could it >>>> be the problem with the command that I am using ? >>>> Many thanks in advance! >>>> >>>> пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>: >>>>> >>>>> Thank you very much, Elaine! >>>>> Indeed, I found that these two options influence the results. >>>>> Cheers, >>>>> Enrico >>>>> >>>>> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: >>>>>> >>>>>> Hello, >>>>>> Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein. >>>>>> >>>>>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> >>>>>> >>>>>> Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive. >>>>>> >>>>>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> >>>>>> >>>>>> I hope this helps, >>>>>> Elaine >>>>>> ----- >>>>>> Elaine C. Meng, Ph.D. >>>>>> UCSF Chimera(X) team >>>>>> Department of Pharmaceutical Chemistry >>>>>> University of California, San Francisco >>>>>> >>>>>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >>>>>>> >>>>>>> Dear ChimeraX users! >>>>>>> I am using the following command to calculate and save in the log >>>>>>> information regarding hydrogen bonds based on the consideration of >>>>>>> multi-frame pdb of the complex >>>>>>> >>>>>>> # calculate hydrogen bonds for the first 14 frames of pdb >>>>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>>>>>> interModel false makePseudobonds false log true intraRes false >>>>>>> saveFile log.txt >>>>>>> >>>>>>> which gives me something like this: >>>>>>> structure_name: >>>>>>> 18 H-bonds >>>>>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >>>>>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 >>>>>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 >>>>>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 >>>>>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 >>>>>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 >>>>>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 >>>>>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 >>>>>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 >>>>>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 >>>>>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 >>>>>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 >>>>>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 >>>>>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A >>>>>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 >>>>>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 >>>>>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 >>>>>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 >>>>>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 >>>>>>>
>>>>>>> >>>>>>> I noticed that in the log there is always information regarding >>>>>>> interactions between the ligand and the side chains of the protein but >>>>>>> nothing regarding hydrogen bonds involved backbone of the protein >>>>>>> (Which is confirmed by X-ray data for my complex). for the test I used >>>>>>> py@ol to visualise h-bonds and may see in the 12th frame the hydrogen >>>>>>> bond involved backbone of the protein.. How could I modify my script >>>>>>> to consider additional hydrogen bonds involving backbone atoms ? >>>>>>> Thank you very much in advance! >>>>>>> Cheers >>>>>>> Enrico >>>>>> >>>> >>> >>> >>> _______________________________________________ >>> ChimeraX-users mailing list >>> ChimeraX-users@cgl.ucsf.edu >>> Manage subscription: >>> https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >>> >> > <test_hbonds-min.png>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi Enrico, That sounds similar to what I did with ProDy then (except I put them at the beginning of each model). When I opened the resulting PDB in ChimeraX then the side chain atoms were disconnected and the backbone maybe as well. After opening in PyMOL first and saving again it was reordered well and they seemed to be connected up right in ChimeraX too. If it's still not working by doing that then you probably need to pass each model to some tool like MODELLER or HADDOCK individually to fix the incorrect bond distances etc. Best wishes James Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hey James, actually, it seems to me that I have already done it: copy/past a receptor model (with the static residues!) into EACH of the model containing docking pose of the ligand + 5 flexible residues. Here is my script to produce multi-model pdb with the complex:
awk 'NR==FNR {s = s $0 ORS; next} $0 == "ENDMDL" {$0 = s $0} 1' "${temp}"/static_receptor.pdb "${results}"/docking_poses_with_flexible_residues.pdb >> "${results}"/complex. pdb
basically it looks for the ENDMDL term in each frame of the multi-model pdb and past there the model of the receptor. So the both are present in each frames.
The issue is related to the Flexible residues, which are always near the ligand atoms (since they are present in the each model with the docking poses) so the resulted "combined" PDB appeared to be "fragmented" in the sence of the side-chains of the flexible residues (taken from the first pdb) as well as their backbone atoms (present in another pdb with the static receptor), so ..
Although the combined PDB produced by my script seems to be absolutely normal, there are problems with the recognition of the back-bone atoms of the flexible residues, which are not considered as the donors for the hydrogen bonds ...
may-be there is some program which may just open my pdb and convert it (the order of the string) into the normal format since the problem is always related to the position of the flexible side-chains in each frame...
Enrico
вт, 22 мар. 2022 г. в 16:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico, It would probably be better to combine them model by model rather than just cat-ing the two. Probably the ProDy Python API could handle this but I'm not exactly sure. You could try something along the lines of the following. I just made some PDB files that sound like what you described and it worked fine.
from prody import *
atoms1 = parsePDB(pdbfile1) # multi-model will be read as multiple coordinate sets
atoms2 = parsePDB(pdbfile2) # I'm assuming it just has one coordinate set
atoms3 = atoms2.copy()
for i in range(atoms1.numCoordsets()-1): atoms3.addCoordset(atoms2)
atoms4 = atoms1 + atoms3
writePDB(pdbfile3, atoms4)
You'd also need to open the resulting PDB file in pymol and save it again (making sure to include all states) to get the atoms back in the right order for chimerax to know they belong to the save residues.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hi James, many thanks for these suggestions! Indeed I am looking for some utility that may be useful to fix my "fragmented" multi-model pdb, converting it into "common" format (considering all frames). I've just made a quick test excluding the residues where I had difficulties with the visualisation of the "back-bone" hydrogen bond from the list of the "flexible residues" in VINA calculations and here it is: since this part is no more "fragmented", Chimera is able to find the hydrogen bond with the default "slopes" values. Victory! May be there is some command in Chimera that may fix the problem? Essentially I am dealing with protein-ligand complex produced by the CAT of two initial pdbs: i) multi-model pdb of the docking poses (containing ligand and 4-5 side-chains) + the "static part" of the protein (all of the atoms excluding these 4-5 flexible side-chains) merged into the each model. It looks absolutely correctly while examining it in any molecular vizualisator so the problem could only be detected in ChimeraX.. I would be grateful for any suggestions Cheers, Enrico
вт, 22 мар. 2022 г. в 12:54, JAMES MICHAEL S1JJRUdFUiA= via ChimeraX-users <chimerax-users@cgl.ucsf.edu>:
Hi Enrico, You may want to try rebuilding these parts with modeller or some other template-based modelling method. There may also be an option to refine the docked structure in autodock like there is in HADDOCK or an ability to use HADDOCK for the autodock output. Best wishes James
Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu>
escribió:
Right, thank you very much Elaine! Now I gotcha! Indeed it should be the case since the pdb was produced by the concatenation of the autodock results, (which left flexible residues with the ligand) with the receptor pdb w/o these side-chains. BTW I double checked and could reproduce the hydrogen bond with the side-chain atoms of the same residue, so the problem indeed is related only to backbone (particularly to the N atom).. Anyway thank you very much for the help! Cheers, Enrico
пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>:
The problem is that your PDB file is fragmented, i.e. you only have some of the receptor residues so the backbone chain is broken in several places. When I try to run "hbonds" on what you sent me, there is a warning message in the Log:
The following atoms were skipped as donors/acceptors due to missing heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; /? ASN 142 N; /? GLN 189 N
When (for example) Glu 166 atom N does not have a bond from residue 165 atom C (because residue 165 is not in your structure), the correct angle for H-bonding is undetermined, so the hbonds algorithm ignores it. If you had the whole protein in the file, it would probably not be missing all these backbone bonds, and then it could correctly detect the H-bonds.
Other programs probably have simple distance checking that doesn't use other atoms to figure out the proper angle.
Whenever you get an unexpected result from a script, it is useful to try it interactively in the GUI so that you can see if there are any warning messages.
I hope this clarifies the situation, Elaine
> On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: > > Hello, > We can't figure out the reason for the "missing" H-bond unless you send us the file with the ligand and receptor atomic coordinates, the same ones shown in the image.. If you didn't want to share it with the whole chimerax-users list but you are willing to share it with the ChimeraX team, you can send it to just my individual e-mail address. However, if you need to keep it completely private we understand. > Best, > Elaine > ----- > Elaine C. Meng, Ph.D. > UCSF Chimera(X) team > Department of Pharmaceutical Chemistry > University of California, San Francisco > >> On Mar 21, 2022, at 2:28 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote: >> >> Hello Elaine >> Many thanks for these kind suggestions! >> In fact this back-bone H-bond has been validated by X-ray data >> (observed in several structures of my protein) and then by
my docking
>> studies. Please find enclosed the picture of this interaction: as we >> may see it is located on the relatively short distance between the O >> atom of the ligand and the HN of the backbone of the Glu residue. I >> believe it has to match both default slope criteria.... >> >> Regarding my command, actually I used it with batch version of >> chimeraX (in the script) so the goal was not to display the >> interactions but rather to save it directly into the log file: >> >> # calculate h-bonds between first 10 models from multi-model pdb file >> hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false >> interModel false makePseudobonds false log true intraRes false >> saveFile log_hbondsALL.log >> >> It produces a log with the correct H-bonds with the exemption of the >> interaction shown on the screenshot (which is always detected by other >> programs...) >> With best regards, >> Enrico >> >> вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>: >>> >>> I see you are not displaying the H-bonds in your command anyway, but another thing that confuses some people is that even though the H-bond is found (i.e. it is counted and listed in the Log) it is not displayed because the atoms are not displayed. >>> >>> In other words, if you are using the display to judge whether the H-bond is found you would need to use the "hbond" command options: >>> >>> makePseudobond true reveal true >>> >>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> >>> >>> Elaine >>> >>> >>>> On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >>>> >>>> Those "slop" values are tolerances, not the scope (cutoff): i.e. they are added to increase the allowed ranges of values for the specific types of atoms. A distSlop of 0.8 does not mean distance cutoff 0.8, it means 0.8 + the strict cutoff (which might be 3.0 or something like that, for a total of 3.8). >>>> >>>> Reasons to not find the H-bond: >>>> >>>> - maybe your atom specification is wrong... in fact I have no idea what the "}" are in your command, they look wrong. However, that would probably cause an error message and you would not get any H-bonds at all. Since you are getting some H-bonds maybe that is not the problem. >>>> >>>> - maybe those types of atoms are not considered by the H-bond detection. I.e. it does not consider C to be an H-bonding type of atom. >>>> >>>> - maybe it is because the H-bond geometry is very poor, you still need to increase the slop values even more to find it. >>>> >>>> However: We believe that Chimera and ChimeraX provide high-quality H-bond detection with the default parameters (or with small increases in the distSlop and angleSlop), so it is unclear why you think your other program finding the H-bond is more correct. Maybe it should not be considered an H-bond. >>>> >>>> Best, >>>> Elaine >>>> ----- >>>> Elaine C. Meng, Ph.D. >>>> UCSF Chimera(X) team >>>> Department of Pharmaceutical Chemistry >>>> University of California, San Francisco >>>> >>>>> On Mar 15, 2022, at 8:17 AM, Enrico Martinez <jmsstarlight@gmail.com> wrote: >>>>> >>>>> Hello again! >>>>> I've just make a test for several docking poses and I confirm that the >>>>> hydrogen bond between ligand and backbone atoms never could be >>>>> detected by chimera using >>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>>>> interModel false makePseudobonds false log true intraRes false >>>>> saveFile log.txt >>>>> >>>>> I tried to increase significantly the distance scope to 0.8 and the >>>>> angle scope to 40 but the interactions could not be detected. Could it >>>>> be the problem with the command that I am using ? >>>>> Many thanks in advance! >>>>> >>>>> пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>: >>>>>> >>>>>> Thank you very much, Elaine! >>>>>> Indeed, I found that these two options influence the results. >>>>>> Cheers, >>>>>> Enrico >>>>>> >>>>>> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: >>>>>>> >>>>>>> Hello, >>>>>>> Specifications like "#1.1&protein" already include the backbone atoms -- they are part of the protein. >>>>>>> >>>>>>>
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
>>>>>>> >>>>>>> Maybe the H-bond(s) that you think should be found are less favorable (longer distance and/or poorer angle). In that case you could try using larger values than the defaults with the "distSlop" and/or "angleSlop" options of "hbonds" to make detection more permissive. >>>>>>> >>>>>>>
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
>>>>>>> >>>>>>> I hope this helps, >>>>>>> Elaine >>>>>>> ----- >>>>>>> Elaine C. Meng, Ph.D. >>>>>>> UCSF Chimera(X) team >>>>>>> Department of Pharmaceutical Chemistry >>>>>>> University of California, San Francisco >>>>>>> >>>>>>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >>>>>>>> >>>>>>>> Dear ChimeraX users! >>>>>>>> I am using the following command to calculate and save in the log >>>>>>>> information regarding hydrogen bonds based on the consideration of >>>>>>>> multi-frame pdb of the complex >>>>>>>> >>>>>>>> # calculate hydrogen bonds for the first 14 frames of pdb >>>>>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >>>>>>>> interModel false makePseudobonds false log true intraRes false >>>>>>>> saveFile log.txt >>>>>>>> >>>>>>>> which gives me something like this: >>>>>>>> structure_name: >>>>>>>> 18 H-bonds >>>>>>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >>>>>>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 2HD2 3.313 2.522 >>>>>>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 H 2.953 2.224 >>>>>>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H 2.753 1.877 >>>>>>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 2HD2 3.240 2.429 >>>>>>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H 3.317 2.389 >>>>>>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 H 3.005 2.272 >>>>>>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H 2.738 2.098 >>>>>>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H 3.054 2.250 >>>>>>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H 3.172 2.528 >>>>>>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 HG 3.828 2.949 >>>>>>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H 3.201 2.393 >>>>>>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 H 3.060 2.238 >>>>>>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen 3.084 N/A >>>>>>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 1HE2 3.148 2.139 >>>>>>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN 189 1HE2 2.941 2.289 >>>>>>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 H 2.985 2.213 >>>>>>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN 189 1HE2 2.803 1.910 >>>>>>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN 142 2HD2 3.199 2.369 >>>>>>>>
>>>>>>>> >>>>>>>> I noticed that in the log there is always information regarding >>>>>>>> interactions between the ligand and the side chains of the protein but >>>>>>>> nothing regarding hydrogen bonds involved backbone of the protein >>>>>>>> (Which is confirmed by X-ray data for my complex). for the test I used >>>>>>>> py@ol to visualise h-bonds and may see in the 12th frame the hydrogen >>>>>>>> bond involved backbone of the protein.. How could I modify my script >>>>>>>> to consider additional hydrogen bonds involving backbone atoms ? >>>>>>>> Thank you very much in advance! >>>>>>>> Cheers >>>>>>>> Enrico >>>>>>> >>>>> >>>> >>>> >>>> _______________________________________________ >>>> ChimeraX-users mailing list >>>> ChimeraX-users@cgl.ucsf.edu >>>> Manage subscription: >>>> https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >>>> >>> >> <test_hbonds-min.png> > > > _______________________________________________ > ChimeraX-users mailing list > ChimeraX-users@cgl.ucsf.edu > Manage subscription: > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Yep, exactly, this is what I may see with my pdb.. BTW do we have some command line solution to "reorder" the atoms in multi-model pdb which me may do the trick "on the fly" via some software executed in the terminal command line (without GUI)? Cheers, Enrico вт, 22 мар. 2022 г. в 17:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico,
That sounds similar to what I did with ProDy then (except I put them at the beginning of each model). When I opened the resulting PDB in ChimeraX then the side chain atoms were disconnected and the backbone maybe as well. After opening in PyMOL first and saving again it was reordered well and they seemed to be connected up right in ChimeraX too. If it's still not working by doing that then you probably need to pass each model to some tool like MODELLER or HADDOCK individually to fix the incorrect bond distances etc.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hey James, actually, it seems to me that I have already done it: copy/past a receptor model (with the static residues!) into EACH of the model containing docking pose of the ligand + 5 flexible residues. Here is my script to produce multi-model pdb with the complex:
awk 'NR==FNR {s = s $0 ORS; next} $0 == "ENDMDL" {$0 = s $0} 1' "${temp}"/static_receptor.pdb "${results}"/docking_poses_with_flexible_residues.pdb >> "${results}"/complex. pdb
basically it looks for the ENDMDL term in each frame of the multi-model pdb and past there the model of the receptor. So the both are present in each frames.
The issue is related to the Flexible residues, which are always near the ligand atoms (since they are present in the each model with the docking poses) so the resulted "combined" PDB appeared to be "fragmented" in the sence of the side-chains of the flexible residues (taken from the first pdb) as well as their backbone atoms (present in another pdb with the static receptor), so ..
Although the combined PDB produced by my script seems to be absolutely normal, there are problems with the recognition of the back-bone atoms of the flexible residues, which are not considered as the donors for the hydrogen bonds ...
may-be there is some program which may just open my pdb and convert it (the order of the string) into the normal format since the problem is always related to the position of the flexible side-chains in each frame...
Enrico
вт, 22 мар. 2022 г. в 16:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico, It would probably be better to combine them model by model rather than just cat-ing the two. Probably the ProDy Python API could handle this but I'm not exactly sure. You could try something along the lines of the following. I just made some PDB files that sound like what you described and it worked fine.
from prody import *
atoms1 = parsePDB(pdbfile1) # multi-model will be read as multiple coordinate sets
atoms2 = parsePDB(pdbfile2) # I'm assuming it just has one coordinate set
atoms3 = atoms2.copy()
for i in range(atoms1.numCoordsets()-1): atoms3.addCoordset(atoms2)
atoms4 = atoms1 + atoms3
writePDB(pdbfile3, atoms4)
You'd also need to open the resulting PDB file in pymol and save it again (making sure to include all states) to get the atoms back in the right order for chimerax to know they belong to the save residues.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hi James, many thanks for these suggestions! Indeed I am looking for some utility that may be useful to fix my "fragmented" multi-model pdb, converting it into "common" format (considering all frames). I've just made a quick test excluding the residues where I had difficulties with the visualisation of the "back-bone" hydrogen bond from the list of the "flexible residues" in VINA calculations and here it is: since this part is no more "fragmented", Chimera is able to find the hydrogen bond with the default "slopes" values. Victory! May be there is some command in Chimera that may fix the problem? Essentially I am dealing with protein-ligand complex produced by the CAT of two initial pdbs: i) multi-model pdb of the docking poses (containing ligand and 4-5 side-chains) + the "static part" of the protein (all of the atoms excluding these 4-5 flexible side-chains) merged into the each model. It looks absolutely correctly while examining it in any molecular vizualisator so the problem could only be detected in ChimeraX.. I would be grateful for any suggestions Cheers, Enrico
вт, 22 мар. 2022 г. в 12:54, JAMES MICHAEL S1JJRUdFUiA= via ChimeraX-users <chimerax-users@cgl.ucsf.edu>:
Hi Enrico, You may want to try rebuilding these parts with modeller or some other template-based modelling method. There may also be an option to refine the docked structure in autodock like there is in HADDOCK or an ability to use HADDOCK for the autodock output. Best wishes James
Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu>
escribió:
Right, thank you very much Elaine! Now I gotcha! Indeed it should be the case since the pdb was produced by the concatenation of the autodock results, (which left flexible residues with the ligand) with the receptor pdb w/o these side-chains. BTW I double checked and could reproduce the hydrogen bond with the side-chain atoms of the same residue, so the problem indeed is related only to backbone (particularly to the N atom).. Anyway thank you very much for the help! Cheers, Enrico
пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>: > > The problem is that your PDB file is fragmented, i.e. you only have > some of the receptor residues so the backbone chain is broken in > several places. When I try to run "hbonds" on what you sent me, > there is a warning message in the Log: > > The following atoms were skipped as donors/acceptors due to missing > heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; > /? ASN 142 N; /? GLN 189 N > > When (for example) Glu 166 atom N does not have a bond from residue > 165 atom C (because residue 165 is not in your structure), the > correct angle for H-bonding is undetermined, so the hbonds > algorithm ignores it. If you had the whole protein in the file, it > would probably not be missing all these backbone bonds, and then it > could correctly detect the H-bonds. > > Other programs probably have simple distance checking that doesn't > use other atoms to figure out the proper angle. > > Whenever you get an unexpected result from a script, it is useful > to try it interactively in the GUI so that you can see if there are > any warning messages. > > I hope this clarifies the situation, > Elaine > > > On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users > <chimerax-users@cgl.ucsf.edu> wrote: > > > > Hello, > > We can't figure out the reason for the "missing" H-bond unless > you send us the file with the ligand and receptor atomic > coordinates, the same ones shown in the image.. If you didn't want > to share it with the whole chimerax-users list but you are willing > to share it with the ChimeraX team, you can send it to just my > individual e-mail address. However, if you need to keep it > completely private we understand. > > Best, > > Elaine > > ----- > > Elaine C. Meng, Ph.D. > > UCSF Chimera(X) team > > Department of Pharmaceutical Chemistry > > University of California, San Francisco > > > >> On Mar 21, 2022, at 2:28 AM, Enrico Martinez > <jmsstarlight@gmail.com> wrote: > >> > >> Hello Elaine > >> Many thanks for these kind suggestions! > >> In fact this back-bone H-bond has been validated by X-ray data > >> (observed in several structures of my protein) and then by
my docking
> >> studies. Please find enclosed the picture of this interaction: as we > >> may see it is located on the relatively short distance between the O > >> atom of the ligand and the HN of the backbone of the Glu residue. I > >> believe it has to match both default slope criteria.... > >> > >> Regarding my command, actually I used it with batch version of > >> chimeraX (in the script) so the goal was not to display the > >> interactions but rather to save it directly into the log file: > >> > >> # calculate h-bonds between first 10 models from multi-model pdb file > >> hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false > >> interModel false makePseudobonds false log true intraRes false > >> saveFile log_hbondsALL.log > >> > >> It produces a log with the correct H-bonds with the exemption of the > >> interaction shown on the screenshot (which is always detected by other > >> programs...) > >> With best regards, > >> Enrico > >> > >> вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>: > >>> > >>> I see you are not displaying the H-bonds in your command > anyway, but another thing that confuses some people is that even > though the H-bond is found (i.e. it is counted and listed in the > Log) it is not displayed because the atoms are not displayed. > >>> > >>> In other words, if you are using the display to judge whether > the H-bond is found you would need to use the "hbond" command > options: > >>> > >>> makePseudobond true reveal true > >>> > >>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> > >>> > >>> Elaine > >>> > >>> > >>>> On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users > <chimerax-users@cgl.ucsf.edu> wrote: > >>>> > >>>> Those "slop" values are tolerances, not the scope (cutoff): > i.e. they are added to increase the allowed ranges of values for > the specific types of atoms. A distSlop of 0.8 does not mean > distance cutoff 0.8, it means 0.8 + the strict cutoff (which might > be 3.0 or something like that, for a total of 3.8). > >>>> > >>>> Reasons to not find the H-bond: > >>>> > >>>> - maybe your atom specification is wrong... in fact I have no > idea what the "}" are in your command, they look wrong. However, > that would probably cause an error message and you would not get > any H-bonds at all. Since you are getting some H-bonds maybe that > is not the problem. > >>>> > >>>> - maybe those types of atoms are not considered by the H-bond > detection. I.e. it does not consider C to be an H-bonding type of > atom. > >>>> > >>>> - maybe it is because the H-bond geometry is very poor, you > still need to increase the slop values even more to find it. > >>>> > >>>> However: We believe that Chimera and ChimeraX provide > high-quality H-bond detection with the default parameters (or with > small increases in the distSlop and angleSlop), so it is unclear > why you think your other program finding the H-bond is more > correct. Maybe it should not be considered an H-bond. > >>>> > >>>> Best, > >>>> Elaine > >>>> ----- > >>>> Elaine C. Meng, Ph.D. > >>>> UCSF Chimera(X) team > >>>> Department of Pharmaceutical Chemistry > >>>> University of California, San Francisco > >>>> > >>>>> On Mar 15, 2022, at 8:17 AM, Enrico Martinez > <jmsstarlight@gmail.com> wrote: > >>>>> > >>>>> Hello again! > >>>>> I've just make a test for several docking poses and I confirm that the > >>>>> hydrogen bond between ligand and backbone atoms never could be > >>>>> detected by chimera using > >>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false > >>>>> interModel false makePseudobonds false log true intraRes false > >>>>> saveFile log.txt > >>>>> > >>>>> I tried to increase significantly the distance scope to 0.8 and the > >>>>> angle scope to 40 but the interactions could not be detected. Could it > >>>>> be the problem with the command that I am using ? > >>>>> Many thanks in advance! > >>>>> > >>>>> пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>: > >>>>>> > >>>>>> Thank you very much, Elaine! > >>>>>> Indeed, I found that these two options influence the results. > >>>>>> Cheers, > >>>>>> Enrico > >>>>>> > >>>>>> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: > >>>>>>> > >>>>>>> Hello, > >>>>>>> Specifications like "#1.1&protein" already include the > backbone atoms -- they are part of the protein. > >>>>>>> > >>>>>>> > <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> > >>>>>>> > >>>>>>> Maybe the H-bond(s) that you think should be found are less > favorable (longer distance and/or poorer angle). In that case you > could try using larger values than the defaults with the "distSlop" > and/or "angleSlop" options of "hbonds" to make detection more > permissive. > >>>>>>> > >>>>>>> > <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> > >>>>>>> > >>>>>>> I hope this helps, > >>>>>>> Elaine > >>>>>>> ----- > >>>>>>> Elaine C. Meng, Ph.D. > >>>>>>> UCSF Chimera(X) team > >>>>>>> Department of Pharmaceutical Chemistry > >>>>>>> University of California, San Francisco > >>>>>>> > >>>>>>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via > ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: > >>>>>>>> > >>>>>>>> Dear ChimeraX users! > >>>>>>>> I am using the following command to calculate and save in the log > >>>>>>>> information regarding hydrogen bonds based on the consideration of > >>>>>>>> multi-frame pdb of the complex > >>>>>>>> > >>>>>>>> # calculate hydrogen bonds for the first 14 frames of pdb > >>>>>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false > >>>>>>>> interModel false makePseudobonds false log true intraRes false > >>>>>>>> saveFile log.txt > >>>>>>>> > >>>>>>>> which gives me something like this: > >>>>>>>> structure_name: > >>>>>>>> 18 H-bonds > >>>>>>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): > >>>>>>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 > 2HD2 3.313 2.522 > >>>>>>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 > H 2.953 2.224 > >>>>>>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H > 2.753 1.877 > >>>>>>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 > 2HD2 3.240 2.429 > >>>>>>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H > 3.317 2.389 > >>>>>>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 > H 3.005 2.272 > >>>>>>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H > 2.738 2.098 > >>>>>>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H > 3.054 2.250 > >>>>>>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H > 3.172 2.528 > >>>>>>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 > HG 3.828 2.949 > >>>>>>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H > 3.201 2.393 > >>>>>>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 > H 3.060 2.238 > >>>>>>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen > 3.084 N/A > >>>>>>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 > 1HE2 3.148 2.139 > >>>>>>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN > 189 1HE2 2.941 2.289 > >>>>>>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 > H 2.985 2.213 > >>>>>>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN > 189 1HE2 2.803 1.910 > >>>>>>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN > 142 2HD2 3.199 2.369 > >>>>>>>> >
> >>>>>>>> > >>>>>>>> I noticed that in the log there is always information regarding > >>>>>>>> interactions between the ligand and the side chains of the > protein but > >>>>>>>> nothing regarding hydrogen bonds involved backbone of the protein > >>>>>>>> (Which is confirmed by X-ray data for my complex). for the > test I used > >>>>>>>> py@ol to visualise h-bonds and may see in the 12th frame > the hydrogen > >>>>>>>> bond involved backbone of the protein.. How could I modify > my script > >>>>>>>> to consider additional hydrogen bonds involving backbone atoms ? > >>>>>>>> Thank you very much in advance! > >>>>>>>> Cheers > >>>>>>>> Enrico > >>>>>>> > >>>>> > >>>> > >>>> > >>>> _______________________________________________ > >>>> ChimeraX-users mailing list > >>>> ChimeraX-users@cgl.ucsf.edu > >>>> Manage subscription: > >>>> https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users > >>>> > >>> > >> <test_hbonds-min.png> > > > > > > _______________________________________________ > > ChimeraX-users mailing list > > ChimeraX-users@cgl.ucsf.edu > > Manage subscription: > > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users > > >
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
No, sorry. It should be possible to run pymol with a script in that way, but I've never actually done it. Best wishes James Enrico Martinez <jmsstarlight@gmail.com> escribió:
Yep, exactly, this is what I may see with my pdb.. BTW do we have some command line solution to "reorder" the atoms in multi-model pdb which me may do the trick "on the fly" via some software executed in the terminal command line (without GUI)? Cheers, Enrico
вт, 22 мар. 2022 г. в 17:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico,
That sounds similar to what I did with ProDy then (except I put them at the beginning of each model). When I opened the resulting PDB in ChimeraX then the side chain atoms were disconnected and the backbone maybe as well. After opening in PyMOL first and saving again it was reordered well and they seemed to be connected up right in ChimeraX too. If it's still not working by doing that then you probably need to pass each model to some tool like MODELLER or HADDOCK individually to fix the incorrect bond distances etc.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hey James, actually, it seems to me that I have already done it: copy/past a receptor model (with the static residues!) into EACH of the model containing docking pose of the ligand + 5 flexible residues. Here is my script to produce multi-model pdb with the complex:
awk 'NR==FNR {s = s $0 ORS; next} $0 == "ENDMDL" {$0 = s $0} 1' "${temp}"/static_receptor.pdb "${results}"/docking_poses_with_flexible_residues.pdb >> "${results}"/complex. pdb
basically it looks for the ENDMDL term in each frame of the multi-model pdb and past there the model of the receptor. So the both are present in each frames.
The issue is related to the Flexible residues, which are always near the ligand atoms (since they are present in the each model with the docking poses) so the resulted "combined" PDB appeared to be "fragmented" in the sence of the side-chains of the flexible residues (taken from the first pdb) as well as their backbone atoms (present in another pdb with the static receptor), so ..
Although the combined PDB produced by my script seems to be absolutely normal, there are problems with the recognition of the back-bone atoms of the flexible residues, which are not considered as the donors for the hydrogen bonds ...
may-be there is some program which may just open my pdb and convert it (the order of the string) into the normal format since the problem is always related to the position of the flexible side-chains in each frame...
Enrico
вт, 22 мар. 2022 г. в 16:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico, It would probably be better to combine them model by model rather than just cat-ing the two. Probably the ProDy Python API could handle this but I'm not exactly sure. You could try something along the lines of the following. I just made some PDB files that sound like what you described and it worked fine.
from prody import *
atoms1 = parsePDB(pdbfile1) # multi-model will be read as multiple coordinate sets
atoms2 = parsePDB(pdbfile2) # I'm assuming it just has one coordinate set
atoms3 = atoms2.copy()
for i in range(atoms1.numCoordsets()-1): atoms3.addCoordset(atoms2)
atoms4 = atoms1 + atoms3
writePDB(pdbfile3, atoms4)
You'd also need to open the resulting PDB file in pymol and save it again (making sure to include all states) to get the atoms back in the right order for chimerax to know they belong to the save residues.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hi James, many thanks for these suggestions! Indeed I am looking for some utility that may be useful to fix my "fragmented" multi-model pdb, converting it into "common" format (considering all frames). I've just made a quick test excluding the residues where I had difficulties with the visualisation of the "back-bone" hydrogen bond from the list of the "flexible residues" in VINA calculations and here it is: since this part is no more "fragmented", Chimera is able to find the hydrogen bond with the default "slopes" values. Victory! May be there is some command in Chimera that may fix the problem? Essentially I am dealing with protein-ligand complex produced by the CAT of two initial pdbs: i) multi-model pdb of the docking poses (containing ligand and 4-5 side-chains) + the "static part" of the protein (all of the atoms excluding these 4-5 flexible side-chains) merged into the each model. It looks absolutely correctly while examining it in any molecular vizualisator so the problem could only be detected in ChimeraX.. I would be grateful for any suggestions Cheers, Enrico
вт, 22 мар. 2022 г. в 12:54, JAMES MICHAEL S1JJRUdFUiA= via ChimeraX-users <chimerax-users@cgl.ucsf.edu>:
Hi Enrico, You may want to try rebuilding these parts with modeller or some other template-based modelling method. There may also be an option to refine the docked structure in autodock like there is in HADDOCK or an ability to use HADDOCK for the autodock output. Best wishes James
Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu>
escribió:
> Right, thank you very much Elaine! > Now I gotcha! Indeed it should be the case since the pdb
was produced
> by the concatenation of the autodock results, (which left flexible > residues with the ligand) with the receptor pdb w/o these side-chains. > BTW I double checked and could reproduce the hydrogen bond with the > side-chain atoms of the same residue, so the problem indeed is related > only to backbone (particularly to the N atom).. > Anyway thank you very much for the help! > Cheers, > Enrico > > пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>: >> >> The problem is that your PDB file is fragmented, i.e. you only have >> some of the receptor residues so the backbone chain is broken in >> several places. When I try to run "hbonds" on what you sent me, >> there is a warning message in the Log: >> >> The following atoms were skipped as donors/acceptors due to missing >> heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; >> /? ASN 142 N; /? GLN 189 N >> >> When (for example) Glu 166 atom N does not have a bond from residue >> 165 atom C (because residue 165 is not in your structure), the >> correct angle for H-bonding is undetermined, so the hbonds >> algorithm ignores it. If you had the whole protein in the file, it >> would probably not be missing all these backbone bonds, and then it >> could correctly detect the H-bonds. >> >> Other programs probably have simple distance checking that doesn't >> use other atoms to figure out the proper angle. >> >> Whenever you get an unexpected result from a script, it is useful >> to try it interactively in the GUI so that you can see if there are >> any warning messages. >> >> I hope this clarifies the situation, >> Elaine >> >> > On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users >> <chimerax-users@cgl.ucsf.edu> wrote: >> > >> > Hello, >> > We can't figure out the reason for the "missing" H-bond unless >> you send us the file with the ligand and receptor atomic >> coordinates, the same ones shown in the image.. If you didn't want >> to share it with the whole chimerax-users list but you are willing >> to share it with the ChimeraX team, you can send it to just my >> individual e-mail address. However, if you need to keep it >> completely private we understand. >> > Best, >> > Elaine >> > ----- >> > Elaine C. Meng, Ph.D. >> > UCSF Chimera(X) team >> > Department of Pharmaceutical Chemistry >> > University of California, San Francisco >> > >> >> On Mar 21, 2022, at 2:28 AM, Enrico Martinez >> <jmsstarlight@gmail.com> wrote: >> >> >> >> Hello Elaine >> >> Many thanks for these kind suggestions! >> >> In fact this back-bone H-bond has been validated by X-ray data >> >> (observed in several structures of my protein) and then by my docking >> >> studies. Please find enclosed the picture of this interaction: as we >> >> may see it is located on the relatively short distance between the O >> >> atom of the ligand and the HN of the backbone of the Glu residue. I >> >> believe it has to match both default slope criteria.... >> >> >> >> Regarding my command, actually I used it with batch version of >> >> chimeraX (in the script) so the goal was not to display the >> >> interactions but rather to save it directly into the log file: >> >> >> >> # calculate h-bonds between first 10 models from multi-model pdb file >> >> hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false >> >> interModel false makePseudobonds false log true intraRes false >> >> saveFile log_hbondsALL.log >> >> >> >> It produces a log with the correct H-bonds with the exemption of the >> >> interaction shown on the screenshot (which is always detected by other >> >> programs...) >> >> With best regards, >> >> Enrico >> >> >> >> вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>: >> >>> >> >>> I see you are not displaying the H-bonds in your command >> anyway, but another thing that confuses some people is that even >> though the H-bond is found (i.e. it is counted and listed in the >> Log) it is not displayed because the atoms are not displayed. >> >>> >> >>> In other words, if you are using the display to judge whether >> the H-bond is found you would need to use the "hbond" command >> options: >> >>> >> >>> makePseudobond true reveal true >> >>> >> >>>
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
>> >>> >> >>> Elaine >> >>> >> >>> >> >>>> On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users >> <chimerax-users@cgl.ucsf.edu> wrote: >> >>>> >> >>>> Those "slop" values are tolerances, not the scope (cutoff): >> i.e. they are added to increase the allowed ranges of values for >> the specific types of atoms. A distSlop of 0.8 does not mean >> distance cutoff 0.8, it means 0.8 + the strict cutoff (which might >> be 3.0 or something like that, for a total of 3.8). >> >>>> >> >>>> Reasons to not find the H-bond: >> >>>> >> >>>> - maybe your atom specification is wrong... in fact I have no >> idea what the "}" are in your command, they look wrong. However, >> that would probably cause an error message and you would not get >> any H-bonds at all. Since you are getting some H-bonds maybe that >> is not the problem. >> >>>> >> >>>> - maybe those types of atoms are not considered by the H-bond >> detection. I.e. it does not consider C to be an H-bonding type of >> atom. >> >>>> >> >>>> - maybe it is because the H-bond geometry is very poor, you >> still need to increase the slop values even more to find it. >> >>>> >> >>>> However: We believe that Chimera and ChimeraX provide >> high-quality H-bond detection with the default parameters (or with >> small increases in the distSlop and angleSlop), so it is unclear >> why you think your other program finding the H-bond is more >> correct. Maybe it should not be considered an H-bond. >> >>>> >> >>>> Best, >> >>>> Elaine >> >>>> ----- >> >>>> Elaine C. Meng, Ph.D. >> >>>> UCSF Chimera(X) team >> >>>> Department of Pharmaceutical Chemistry >> >>>> University of California, San Francisco >> >>>> >> >>>>> On Mar 15, 2022, at 8:17 AM, Enrico Martinez >> <jmsstarlight@gmail.com> wrote: >> >>>>> >> >>>>> Hello again! >> >>>>> I've just make a test for several docking poses and I confirm that the >> >>>>> hydrogen bond between ligand and backbone atoms never could be >> >>>>> detected by chimera using >> >>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >> >>>>> interModel false makePseudobonds false log true intraRes false >> >>>>> saveFile log.txt >> >>>>> >> >>>>> I tried to increase significantly the distance scope to 0.8 and the >> >>>>> angle scope to 40 but the interactions could not be detected. Could it >> >>>>> be the problem with the command that I am using ? >> >>>>> Many thanks in advance! >> >>>>> >> >>>>> пн, 14 мар. 2022 г. в 15:38, Enrico Martinez <jmsstarlight@gmail.com>: >> >>>>>> >> >>>>>> Thank you very much, Elaine! >> >>>>>> Indeed, I found that these two options influence the results. >> >>>>>> Cheers, >> >>>>>> Enrico >> >>>>>> >> >>>>>> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: >> >>>>>>> >> >>>>>>> Hello, >> >>>>>>> Specifications like "#1.1&protein" already include the >> backbone atoms -- they are part of the protein. >> >>>>>>> >> >>>>>>> >> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> >> >>>>>>> >> >>>>>>> Maybe the H-bond(s) that you think should be found are less >> favorable (longer distance and/or poorer angle). In that case you >> could try using larger values than the defaults with the "distSlop" >> and/or "angleSlop" options of "hbonds" to make detection more >> permissive. >> >>>>>>> >> >>>>>>> >> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> >> >>>>>>> >> >>>>>>> I hope this helps, >> >>>>>>> Elaine >> >>>>>>> ----- >> >>>>>>> Elaine C. Meng, Ph.D. >> >>>>>>> UCSF Chimera(X) team >> >>>>>>> Department of Pharmaceutical Chemistry >> >>>>>>> University of California, San Francisco >> >>>>>>> >> >>>>>>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via >> ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >> >>>>>>>> >> >>>>>>>> Dear ChimeraX users! >> >>>>>>>> I am using the following command to calculate and save in the log >> >>>>>>>> information regarding hydrogen bonds based on the consideration of >> >>>>>>>> multi-frame pdb of the complex >> >>>>>>>> >> >>>>>>>> # calculate hydrogen bonds for the first 14 frames of pdb >> >>>>>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >> >>>>>>>> interModel false makePseudobonds false log true intraRes false >> >>>>>>>> saveFile log.txt >> >>>>>>>> >> >>>>>>>> which gives me something like this: >> >>>>>>>> structure_name: >> >>>>>>>> 18 H-bonds >> >>>>>>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >> >>>>>>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 >> 2HD2 3.313 2.522 >> >>>>>>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 >> H 2.953 2.224 >> >>>>>>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H >> 2.753 1.877 >> >>>>>>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 >> 2HD2 3.240 2.429 >> >>>>>>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H >> 3.317 2.389 >> >>>>>>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 >> H 3.005 2.272 >> >>>>>>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H >> 2.738 2.098 >> >>>>>>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H >> 3.054 2.250 >> >>>>>>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H >> 3.172 2.528 >> >>>>>>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 >> HG 3.828 2.949 >> >>>>>>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H >> 3.201 2.393 >> >>>>>>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 >> H 3.060 2.238 >> >>>>>>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen >> 3.084 N/A >> >>>>>>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 >> 1HE2 3.148 2.139 >> >>>>>>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN >> 189 1HE2 2.941 2.289 >> >>>>>>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 >> H 2.985 2.213 >> >>>>>>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN >> 189 1HE2 2.803 1.910 >> >>>>>>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN >> 142 2HD2 3.199 2.369 >> >>>>>>>> >>
>> >>>>>>>> >> >>>>>>>> I noticed that in the log there is always information regarding >> >>>>>>>> interactions between the ligand and the side chains of the >> protein but >> >>>>>>>> nothing regarding hydrogen bonds involved backbone of the protein >> >>>>>>>> (Which is confirmed by X-ray data for my complex). for the >> test I used >> >>>>>>>> py@ol to visualise h-bonds and may see in the 12th frame >> the hydrogen >> >>>>>>>> bond involved backbone of the protein.. How could I modify >> my script >> >>>>>>>> to consider additional hydrogen bonds involving backbone atoms ? >> >>>>>>>> Thank you very much in advance! >> >>>>>>>> Cheers >> >>>>>>>> Enrico >> >>>>>>> >> >>>>> >> >>>> >> >>>> >> >>>> _______________________________________________ >> >>>> ChimeraX-users mailing list >> >>>> ChimeraX-users@cgl.ucsf.edu >> >>>> Manage subscription: >> >>>> https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >> >>>> >> >>> >> >> <test_hbonds-min.png> >> > >> > >> > _______________________________________________ >> > ChimeraX-users mailing list >> > ChimeraX-users@cgl.ucsf.edu >> > Manage subscription: >> > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >> > >> > > _______________________________________________ > ChimeraX-users mailing list > ChimeraX-users@cgl.ucsf.edu > Manage subscription: > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Right, thank you James! Indeed the following script using pymol directly in bash could fix the issue with my pdb so the ChimeraX recognize all hydrogen bonds correctly! #!/usr/local/bin/bash home="$PWD" pdb="${home}"/input.pdb #run pymol in batch mode to fix the pdb pymol -c -d " from pymol import cmd cmd.load('$pdb') # execute some command to fix atom order in the multi-model pdb cmd.save('input_proc.pdb', state='0') " вт, 22 мар. 2022 г. в 18:04, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
No, sorry. It should be possible to run pymol with a script in that way, but I've never actually done it. Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Yep, exactly, this is what I may see with my pdb.. BTW do we have some command line solution to "reorder" the atoms in multi-model pdb which me may do the trick "on the fly" via some software executed in the terminal command line (without GUI)? Cheers, Enrico
вт, 22 мар. 2022 г. в 17:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico,
That sounds similar to what I did with ProDy then (except I put them at the beginning of each model). When I opened the resulting PDB in ChimeraX then the side chain atoms were disconnected and the backbone maybe as well. After opening in PyMOL first and saving again it was reordered well and they seemed to be connected up right in ChimeraX too. If it's still not working by doing that then you probably need to pass each model to some tool like MODELLER or HADDOCK individually to fix the incorrect bond distances etc.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hey James, actually, it seems to me that I have already done it: copy/past a receptor model (with the static residues!) into EACH of the model containing docking pose of the ligand + 5 flexible residues. Here is my script to produce multi-model pdb with the complex:
awk 'NR==FNR {s = s $0 ORS; next} $0 == "ENDMDL" {$0 = s $0} 1' "${temp}"/static_receptor.pdb "${results}"/docking_poses_with_flexible_residues.pdb >> "${results}"/complex. pdb
basically it looks for the ENDMDL term in each frame of the multi-model pdb and past there the model of the receptor. So the both are present in each frames.
The issue is related to the Flexible residues, which are always near the ligand atoms (since they are present in the each model with the docking poses) so the resulted "combined" PDB appeared to be "fragmented" in the sence of the side-chains of the flexible residues (taken from the first pdb) as well as their backbone atoms (present in another pdb with the static receptor), so ..
Although the combined PDB produced by my script seems to be absolutely normal, there are problems with the recognition of the back-bone atoms of the flexible residues, which are not considered as the donors for the hydrogen bonds ...
may-be there is some program which may just open my pdb and convert it (the order of the string) into the normal format since the problem is always related to the position of the flexible side-chains in each frame...
Enrico
вт, 22 мар. 2022 г. в 16:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico, It would probably be better to combine them model by model rather than just cat-ing the two. Probably the ProDy Python API could handle this but I'm not exactly sure. You could try something along the lines of the following. I just made some PDB files that sound like what you described and it worked fine.
from prody import *
atoms1 = parsePDB(pdbfile1) # multi-model will be read as multiple coordinate sets
atoms2 = parsePDB(pdbfile2) # I'm assuming it just has one coordinate set
atoms3 = atoms2.copy()
for i in range(atoms1.numCoordsets()-1): atoms3.addCoordset(atoms2)
atoms4 = atoms1 + atoms3
writePDB(pdbfile3, atoms4)
You'd also need to open the resulting PDB file in pymol and save it again (making sure to include all states) to get the atoms back in the right order for chimerax to know they belong to the save residues.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hi James, many thanks for these suggestions! Indeed I am looking for some utility that may be useful to fix my "fragmented" multi-model pdb, converting it into "common" format (considering all frames). I've just made a quick test excluding the residues where I had difficulties with the visualisation of the "back-bone" hydrogen bond from the list of the "flexible residues" in VINA calculations and here it is: since this part is no more "fragmented", Chimera is able to find the hydrogen bond with the default "slopes" values. Victory! May be there is some command in Chimera that may fix the problem? Essentially I am dealing with protein-ligand complex produced by the CAT of two initial pdbs: i) multi-model pdb of the docking poses (containing ligand and 4-5 side-chains) + the "static part" of the protein (all of the atoms excluding these 4-5 flexible side-chains) merged into the each model. It looks absolutely correctly while examining it in any molecular vizualisator so the problem could only be detected in ChimeraX.. I would be grateful for any suggestions Cheers, Enrico
вт, 22 мар. 2022 г. в 12:54, JAMES MICHAEL S1JJRUdFUiA= via ChimeraX-users <chimerax-users@cgl.ucsf.edu>: > > Hi Enrico, > You may want to try rebuilding these parts with modeller or some other > template-based modelling method. There may also be an option to refine > the docked structure in autodock like there is in HADDOCK or an > ability to use HADDOCK for the autodock output. > Best wishes > James > > Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> escribió: > > > Right, thank you very much Elaine! > > Now I gotcha! Indeed it should be the case since the pdb
was produced
> > by the concatenation of the autodock results, (which left flexible > > residues with the ligand) with the receptor pdb w/o these side-chains. > > BTW I double checked and could reproduce the hydrogen bond with the > > side-chain atoms of the same residue, so the problem indeed is related > > only to backbone (particularly to the N atom).. > > Anyway thank you very much for the help! > > Cheers, > > Enrico > > > > пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>: > >> > >> The problem is that your PDB file is fragmented, i.e. you only have > >> some of the receptor residues so the backbone chain is broken in > >> several places. When I try to run "hbonds" on what you sent me, > >> there is a warning message in the Log: > >> > >> The following atoms were skipped as donors/acceptors due to missing > >> heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; > >> /? ASN 142 N; /? GLN 189 N > >> > >> When (for example) Glu 166 atom N does not have a bond from residue > >> 165 atom C (because residue 165 is not in your structure), the > >> correct angle for H-bonding is undetermined, so the hbonds > >> algorithm ignores it. If you had the whole protein in the file, it > >> would probably not be missing all these backbone bonds, and then it > >> could correctly detect the H-bonds. > >> > >> Other programs probably have simple distance checking that doesn't > >> use other atoms to figure out the proper angle. > >> > >> Whenever you get an unexpected result from a script, it is useful > >> to try it interactively in the GUI so that you can see if there are > >> any warning messages. > >> > >> I hope this clarifies the situation, > >> Elaine > >> > >> > On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users > >> <chimerax-users@cgl.ucsf.edu> wrote: > >> > > >> > Hello, > >> > We can't figure out the reason for the "missing" H-bond unless > >> you send us the file with the ligand and receptor atomic > >> coordinates, the same ones shown in the image.. If you didn't want > >> to share it with the whole chimerax-users list but you are willing > >> to share it with the ChimeraX team, you can send it to just my > >> individual e-mail address. However, if you need to keep it > >> completely private we understand. > >> > Best, > >> > Elaine > >> > ----- > >> > Elaine C. Meng, Ph.D. > >> > UCSF Chimera(X) team > >> > Department of Pharmaceutical Chemistry > >> > University of California, San Francisco > >> > > >> >> On Mar 21, 2022, at 2:28 AM, Enrico Martinez > >> <jmsstarlight@gmail.com> wrote: > >> >> > >> >> Hello Elaine > >> >> Many thanks for these kind suggestions! > >> >> In fact this back-bone H-bond has been validated by X-ray data > >> >> (observed in several structures of my protein) and then by my docking > >> >> studies. Please find enclosed the picture of this interaction: as we > >> >> may see it is located on the relatively short distance between the O > >> >> atom of the ligand and the HN of the backbone of the Glu residue. I > >> >> believe it has to match both default slope criteria.... > >> >> > >> >> Regarding my command, actually I used it with batch version of > >> >> chimeraX (in the script) so the goal was not to display the > >> >> interactions but rather to save it directly into the log file: > >> >> > >> >> # calculate h-bonds between first 10 models from multi-model pdb file > >> >> hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false > >> >> interModel false makePseudobonds false log true intraRes false > >> >> saveFile log_hbondsALL.log > >> >> > >> >> It produces a log with the correct H-bonds with the exemption of the > >> >> interaction shown on the screenshot (which is always detected by other > >> >> programs...) > >> >> With best regards, > >> >> Enrico > >> >> > >> >> вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>: > >> >>> > >> >>> I see you are not displaying the H-bonds in your command > >> anyway, but another thing that confuses some people is that even > >> though the H-bond is found (i.e. it is counted and listed in the > >> Log) it is not displayed because the atoms are not displayed. > >> >>> > >> >>> In other words, if you are using the display to judge whether > >> the H-bond is found you would need to use the "hbond" command > >> options: > >> >>> > >> >>> makePseudobond true reveal true > >> >>> > >> >>> > <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> > >> >>> > >> >>> Elaine > >> >>> > >> >>> > >> >>>> On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users > >> <chimerax-users@cgl.ucsf.edu> wrote: > >> >>>> > >> >>>> Those "slop" values are tolerances, not the scope (cutoff): > >> i.e. they are added to increase the allowed ranges of values for > >> the specific types of atoms. A distSlop of 0.8 does not mean > >> distance cutoff 0.8, it means 0.8 + the strict cutoff (which might > >> be 3.0 or something like that, for a total of 3.8). > >> >>>> > >> >>>> Reasons to not find the H-bond: > >> >>>> > >> >>>> - maybe your atom specification is wrong... in fact I have no > >> idea what the "}" are in your command, they look wrong. However, > >> that would probably cause an error message and you would not get > >> any H-bonds at all. Since you are getting some H-bonds maybe that > >> is not the problem. > >> >>>> > >> >>>> - maybe those types of atoms are not considered by the H-bond > >> detection. I.e. it does not consider C to be an H-bonding type of > >> atom. > >> >>>> > >> >>>> - maybe it is because the H-bond geometry is very poor, you > >> still need to increase the slop values even more to find it. > >> >>>> > >> >>>> However: We believe that Chimera and ChimeraX provide > >> high-quality H-bond detection with the default parameters (or with > >> small increases in the distSlop and angleSlop), so it is unclear > >> why you think your other program finding the H-bond is more > >> correct. Maybe it should not be considered an H-bond. > >> >>>> > >> >>>> Best, > >> >>>> Elaine > >> >>>> ----- > >> >>>> Elaine C. Meng, Ph.D. > >> >>>> UCSF Chimera(X) team > >> >>>> Department of Pharmaceutical Chemistry > >> >>>> University of California, San Francisco > >> >>>> > >> >>>>> On Mar 15, 2022, at 8:17 AM, Enrico Martinez > >> <jmsstarlight@gmail.com> wrote: > >> >>>>> > >> >>>>> Hello again! > >> >>>>> I've just make a test for several docking poses and I > confirm that the > >> >>>>> hydrogen bond between ligand and backbone atoms never could be > >> >>>>> detected by chimera using > >> >>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false > >> >>>>> interModel false makePseudobonds false log true intraRes false > >> >>>>> saveFile log.txt > >> >>>>> > >> >>>>> I tried to increase significantly the distance scope to 0.8 and the > >> >>>>> angle scope to 40 but the interactions could not be > detected. Could it > >> >>>>> be the problem with the command that I am using ? > >> >>>>> Many thanks in advance! > >> >>>>> > >> >>>>> пн, 14 мар. 2022 г. в 15:38, Enrico Martinez > <jmsstarlight@gmail.com>: > >> >>>>>> > >> >>>>>> Thank you very much, Elaine! > >> >>>>>> Indeed, I found that these two options influence the results. > >> >>>>>> Cheers, > >> >>>>>> Enrico > >> >>>>>> > >> >>>>>> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: > >> >>>>>>> > >> >>>>>>> Hello, > >> >>>>>>> Specifications like "#1.1&protein" already include the > >> backbone atoms -- they are part of the protein. > >> >>>>>>> > >> >>>>>>> > >> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> > >> >>>>>>> > >> >>>>>>> Maybe the H-bond(s) that you think should be found are less > >> favorable (longer distance and/or poorer angle). In that case you > >> could try using larger values than the defaults with the "distSlop" > >> and/or "angleSlop" options of "hbonds" to make detection more > >> permissive. > >> >>>>>>> > >> >>>>>>> > >> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> > >> >>>>>>> > >> >>>>>>> I hope this helps, > >> >>>>>>> Elaine > >> >>>>>>> ----- > >> >>>>>>> Elaine C. Meng, Ph.D. > >> >>>>>>> UCSF Chimera(X) team > >> >>>>>>> Department of Pharmaceutical Chemistry > >> >>>>>>> University of California, San Francisco > >> >>>>>>> > >> >>>>>>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via > >> ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: > >> >>>>>>>> > >> >>>>>>>> Dear ChimeraX users! > >> >>>>>>>> I am using the following command to calculate and save > in the log > >> >>>>>>>> information regarding hydrogen bonds based on the > consideration of > >> >>>>>>>> multi-frame pdb of the complex > >> >>>>>>>> > >> >>>>>>>> # calculate hydrogen bonds for the first 14 frames of pdb > >> >>>>>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false > >> >>>>>>>> interModel false makePseudobonds false log true intraRes false > >> >>>>>>>> saveFile log.txt > >> >>>>>>>> > >> >>>>>>>> which gives me something like this: > >> >>>>>>>> structure_name: > >> >>>>>>>> 18 H-bonds > >> >>>>>>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): > >> >>>>>>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 > >> 2HD2 3.313 2.522 > >> >>>>>>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 > >> H 2.953 2.224 > >> >>>>>>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H > >> 2.753 1.877 > >> >>>>>>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 > >> 2HD2 3.240 2.429 > >> >>>>>>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H > >> 3.317 2.389 > >> >>>>>>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 > >> H 3.005 2.272 > >> >>>>>>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H > >> 2.738 2.098 > >> >>>>>>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H > >> 3.054 2.250 > >> >>>>>>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H > >> 3.172 2.528 > >> >>>>>>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 > >> HG 3.828 2.949 > >> >>>>>>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H > >> 3.201 2.393 > >> >>>>>>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 > >> H 3.060 2.238 > >> >>>>>>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen > >> 3.084 N/A > >> >>>>>>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 > >> 1HE2 3.148 2.139 > >> >>>>>>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN > >> 189 1HE2 2.941 2.289 > >> >>>>>>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 > >> H 2.985 2.213 > >> >>>>>>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN > >> 189 1HE2 2.803 1.910 > >> >>>>>>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN > >> 142 2HD2 3.199 2.369 > >> >>>>>>>> > >> >
> >> >>>>>>>> > >> >>>>>>>> I noticed that in the log there is always information regarding > >> >>>>>>>> interactions between the ligand and the side chains of the > >> protein but > >> >>>>>>>> nothing regarding hydrogen bonds involved backbone of > the protein > >> >>>>>>>> (Which is confirmed by X-ray data for my complex). for the > >> test I used > >> >>>>>>>> py@ol to visualise h-bonds and may see in the 12th frame > >> the hydrogen > >> >>>>>>>> bond involved backbone of the protein.. How could I modify > >> my script > >> >>>>>>>> to consider additional hydrogen bonds involving backbone atoms ? > >> >>>>>>>> Thank you very much in advance! > >> >>>>>>>> Cheers > >> >>>>>>>> Enrico > >> >>>>>>> > >> >>>>> > >> >>>> > >> >>>> > >> >>>> _______________________________________________ > >> >>>> ChimeraX-users mailing list > >> >>>> ChimeraX-users@cgl.ucsf.edu > >> >>>> Manage subscription: > >> >>>> https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users > >> >>>> > >> >>> > >> >> <test_hbonds-min.png> > >> > > >> > > >> > _______________________________________________ > >> > ChimeraX-users mailing list > >> > ChimeraX-users@cgl.ucsf.edu > >> > Manage subscription: > >> > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users > >> > > >> > > > > _______________________________________________ > > ChimeraX-users mailing list > > ChimeraX-users@cgl.ucsf.edu > > Manage subscription: > > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users > > > > _______________________________________________ > ChimeraX-users mailing list > ChimeraX-users@cgl.ucsf.edu > Manage subscription: > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Perfect! Thanks for the useful update too! Enrico Martinez <jmsstarlight@gmail.com> escribió:
Right, thank you James! Indeed the following script using pymol directly in bash could fix the issue with my pdb so the ChimeraX recognize all hydrogen bonds correctly!
#!/usr/local/bin/bash home="$PWD" pdb="${home}"/input.pdb
#run pymol in batch mode to fix the pdb pymol -c -d " from pymol import cmd cmd.load('$pdb') # execute some command to fix atom order in the multi-model pdb cmd.save('input_proc.pdb', state='0') "
вт, 22 мар. 2022 г. в 18:04, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
No, sorry. It should be possible to run pymol with a script in that way, but I've never actually done it. Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Yep, exactly, this is what I may see with my pdb.. BTW do we have some command line solution to "reorder" the atoms in multi-model pdb which me may do the trick "on the fly" via some software executed in the terminal command line (without GUI)? Cheers, Enrico
вт, 22 мар. 2022 г. в 17:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico,
That sounds similar to what I did with ProDy then (except I put them at the beginning of each model). When I opened the resulting PDB in ChimeraX then the side chain atoms were disconnected and the backbone maybe as well. After opening in PyMOL first and saving again it was reordered well and they seemed to be connected up right in ChimeraX too. If it's still not working by doing that then you probably need to pass each model to some tool like MODELLER or HADDOCK individually to fix the incorrect bond distances etc.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
Hey James, actually, it seems to me that I have already done it: copy/past a receptor model (with the static residues!) into EACH of the model containing docking pose of the ligand + 5 flexible residues. Here is my script to produce multi-model pdb with the complex:
awk 'NR==FNR {s = s $0 ORS; next} $0 == "ENDMDL" {$0 = s $0} 1' "${temp}"/static_receptor.pdb "${results}"/docking_poses_with_flexible_residues.pdb >> "${results}"/complex. pdb
basically it looks for the ENDMDL term in each frame of the multi-model pdb and past there the model of the receptor. So the both are present in each frames.
The issue is related to the Flexible residues, which are always near the ligand atoms (since they are present in the each model with the docking poses) so the resulted "combined" PDB appeared to be "fragmented" in the sence of the side-chains of the flexible residues (taken from the first pdb) as well as their backbone atoms (present in another pdb with the static receptor), so ..
Although the combined PDB produced by my script seems to be absolutely normal, there are problems with the recognition of the back-bone atoms of the flexible residues, which are not considered as the donors for the hydrogen bonds ...
may-be there is some program which may just open my pdb and convert it (the order of the string) into the normal format since the problem is always related to the position of the flexible side-chains in each frame...
Enrico
вт, 22 мар. 2022 г. в 16:12, JAMES MICHAEL S1JJRUdFUiA= <jmkrieger@cnb.csic.es>:
Hi Enrico, It would probably be better to combine them model by model rather than just cat-ing the two. Probably the ProDy Python API could handle this but I'm not exactly sure. You could try something along the lines of the following. I just made some PDB files that sound like what you described and it worked fine.
from prody import *
atoms1 = parsePDB(pdbfile1) # multi-model will be read as multiple coordinate sets
atoms2 = parsePDB(pdbfile2) # I'm assuming it just has one
coordinate set
atoms3 = atoms2.copy()
for i in range(atoms1.numCoordsets()-1): atoms3.addCoordset(atoms2)
atoms4 = atoms1 + atoms3
writePDB(pdbfile3, atoms4)
You'd also need to open the resulting PDB file in pymol and save it again (making sure to include all states) to get the atoms back in the right order for chimerax to know they belong to the save residues.
Best wishes James
Enrico Martinez <jmsstarlight@gmail.com> escribió:
> Hi James, > many thanks for these suggestions! > Indeed I am looking for some utility that may be useful to fix my > "fragmented" multi-model pdb, converting it into "common" format > (considering all frames). > I've just made a quick test excluding the residues where I had > difficulties with the visualisation of the "back-bone" hydrogen bond > from the list of the "flexible residues" in VINA
calculations and here
> it is: since this part is no more "fragmented", Chimera is able to > find the hydrogen bond with the default "slopes" values. Victory! > May be there is some command in Chimera that may fix the problem? > Essentially I am dealing with protein-ligand complex produced by the > CAT of two initial pdbs: i) multi-model pdb of the docking poses > (containing ligand and 4-5 side-chains) + the "static part" of the > protein (all of the atoms excluding these 4-5 flexible side-chains) > merged into the each model. It looks absolutely correctly while > examining it in any molecular vizualisator so the problem could only > be detected in ChimeraX.. > I would be grateful for any suggestions > Cheers, > Enrico > > вт, 22 мар. 2022 г. в 12:54, JAMES MICHAEL S1JJRUdFUiA= via > ChimeraX-users <chimerax-users@cgl.ucsf.edu>: >> >> Hi Enrico, >> You may want to try rebuilding these parts with modeller or some other >> template-based modelling method. There may also be an option to refine >> the docked structure in autodock like there is in HADDOCK or an >> ability to use HADDOCK for the autodock output. >> Best wishes >> James >> >> Enrico Martinez via ChimeraX-users <chimerax-users@cgl.ucsf.edu> escribió: >> >> > Right, thank you very much Elaine! >> > Now I gotcha! Indeed it should be the case since the pdb was produced >> > by the concatenation of the autodock results, (which left flexible >> > residues with the ligand) with the receptor pdb w/o these side-chains. >> > BTW I double checked and could reproduce the hydrogen bond with the >> > side-chain atoms of the same residue, so the problem indeed is related >> > only to backbone (particularly to the N atom).. >> > Anyway thank you very much for the help! >> > Cheers, >> > Enrico >> > >> > пн, 21 мар. 2022 г. в 19:56, Elaine Meng <meng@cgl.ucsf.edu>: >> >> >> >> The problem is that your PDB file is fragmented, i.e. you only have >> >> some of the receptor residues so the backbone chain is broken in >> >> several places. When I try to run "hbonds" on what you sent me, >> >> there is a warning message in the Log: >> >> >> >> The following atoms were skipped as donors/acceptors due to missing >> >> heavy-atom bond partners: /? MET 49 N; /? GLU 166 N; /? CYS 145 N; >> >> /? ASN 142 N; /? GLN 189 N >> >> >> >> When (for example) Glu 166 atom N does not have a bond from residue >> >> 165 atom C (because residue 165 is not in your structure), the >> >> correct angle for H-bonding is undetermined, so the hbonds >> >> algorithm ignores it. If you had the whole protein in the file, it >> >> would probably not be missing all these backbone bonds, and then it >> >> could correctly detect the H-bonds. >> >> >> >> Other programs probably have simple distance checking that doesn't >> >> use other atoms to figure out the proper angle. >> >> >> >> Whenever you get an unexpected result from a script, it is useful >> >> to try it interactively in the GUI so that you can see if there are >> >> any warning messages. >> >> >> >> I hope this clarifies the situation, >> >> Elaine >> >> >> >> > On Mar 21, 2022, at 9:09 AM, Elaine Meng via ChimeraX-users >> >> <chimerax-users@cgl.ucsf.edu> wrote: >> >> > >> >> > Hello, >> >> > We can't figure out the reason for the "missing" H-bond unless >> >> you send us the file with the ligand and receptor atomic >> >> coordinates, the same ones shown in the image.. If you didn't want >> >> to share it with the whole chimerax-users list but you are willing >> >> to share it with the ChimeraX team, you can send it to just my >> >> individual e-mail address. However, if you need to keep it >> >> completely private we understand. >> >> > Best, >> >> > Elaine >> >> > ----- >> >> > Elaine C. Meng, Ph.D. >> >> > UCSF Chimera(X) team >> >> > Department of Pharmaceutical Chemistry >> >> > University of California, San Francisco >> >> > >> >> >> On Mar 21, 2022, at 2:28 AM, Enrico Martinez >> >> <jmsstarlight@gmail.com> wrote: >> >> >> >> >> >> Hello Elaine >> >> >> Many thanks for these kind suggestions! >> >> >> In fact this back-bone H-bond has been validated by X-ray data >> >> >> (observed in several structures of my protein) and then by my docking >> >> >> studies. Please find enclosed the picture of this interaction: as we >> >> >> may see it is located on the relatively short distance between the O >> >> >> atom of the ligand and the HN of the backbone of the Glu residue. I >> >> >> believe it has to match both default slope criteria.... >> >> >> >> >> >> Regarding my command, actually I used it with batch version of >> >> >> chimeraX (in the script) so the goal was not to display the >> >> >> interactions but rather to save it directly into the log file: >> >> >> >> >> >> # calculate h-bonds between first 10 models from multi-model pdb file >> >> >> hbonds #1.1-10&protein restrict #1.1-10&ligand coordsets false >> >> >> interModel false makePseudobonds false log true intraRes false >> >> >> saveFile log_hbondsALL.log >> >> >> >> >> >> It produces a log with the correct H-bonds with the exemption of the >> >> >> interaction shown on the screenshot (which is always detected by other >> >> >> programs...) >> >> >> With best regards, >> >> >> Enrico >> >> >> >> >> >> вт, 15 мар. 2022 г. в 17:28, Elaine Meng <meng@cgl.ucsf.edu>: >> >> >>> >> >> >>> I see you are not displaying the H-bonds in your command >> >> anyway, but another thing that confuses some people is that even >> >> though the H-bond is found (i.e. it is counted and listed in the >> >> Log) it is not displayed because the atoms are not displayed. >> >> >>> >> >> >>> In other words, if you are using the display to judge whether >> >> the H-bond is found you would need to use the "hbond" command >> >> options: >> >> >>> >> >> >>> makePseudobond true reveal true >> >> >>> >> >> >>> >> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options> >> >> >>> >> >> >>> Elaine >> >> >>> >> >> >>> >> >> >>>> On Mar 15, 2022, at 9:20 AM, Elaine Meng via ChimeraX-users >> >> <chimerax-users@cgl.ucsf.edu> wrote: >> >> >>>> >> >> >>>> Those "slop" values are tolerances, not the scope (cutoff): >> >> i.e. they are added to increase the allowed ranges of values for >> >> the specific types of atoms. A distSlop of 0.8 does not mean >> >> distance cutoff 0.8, it means 0.8 + the strict cutoff (which might >> >> be 3.0 or something like that, for a total of 3.8). >> >> >>>> >> >> >>>> Reasons to not find the H-bond: >> >> >>>> >> >> >>>> - maybe your atom specification is wrong... in fact I have no >> >> idea what the "}" are in your command, they look wrong. However, >> >> that would probably cause an error message and you would not get >> >> any H-bonds at all. Since you are getting some H-bonds maybe that >> >> is not the problem. >> >> >>>> >> >> >>>> - maybe those types of atoms are not considered by the H-bond >> >> detection. I.e. it does not consider C to be an H-bonding type of >> >> atom. >> >> >>>> >> >> >>>> - maybe it is because the H-bond geometry is very poor, you >> >> still need to increase the slop values even more to find it. >> >> >>>> >> >> >>>> However: We believe that Chimera and ChimeraX provide >> >> high-quality H-bond detection with the default parameters (or with >> >> small increases in the distSlop and angleSlop), so it is unclear >> >> why you think your other program finding the H-bond is more >> >> correct. Maybe it should not be considered an H-bond. >> >> >>>> >> >> >>>> Best, >> >> >>>> Elaine >> >> >>>> ----- >> >> >>>> Elaine C. Meng, Ph.D. >> >> >>>> UCSF Chimera(X) team >> >> >>>> Department of Pharmaceutical Chemistry >> >> >>>> University of California, San Francisco >> >> >>>> >> >> >>>>> On Mar 15, 2022, at 8:17 AM, Enrico Martinez >> >> <jmsstarlight@gmail.com> wrote: >> >> >>>>> >> >> >>>>> Hello again! >> >> >>>>> I've just make a test for several docking poses and I >> confirm that the >> >> >>>>> hydrogen bond between ligand and backbone atoms never could be >> >> >>>>> detected by chimera using >> >> >>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >> >> >>>>> interModel false makePseudobonds false log true intraRes false >> >> >>>>> saveFile log.txt >> >> >>>>> >> >> >>>>> I tried to increase significantly the distance scope to 0.8 and the >> >> >>>>> angle scope to 40 but the interactions could not be >> detected. Could it >> >> >>>>> be the problem with the command that I am using ? >> >> >>>>> Many thanks in advance! >> >> >>>>> >> >> >>>>> пн, 14 мар. 2022 г. в 15:38, Enrico Martinez >> <jmsstarlight@gmail.com>: >> >> >>>>>> >> >> >>>>>> Thank you very much, Elaine! >> >> >>>>>> Indeed, I found that these two options influence the results. >> >> >>>>>> Cheers, >> >> >>>>>> Enrico >> >> >>>>>> >> >> >>>>>> чт, 10 мар. 2022 г. в 19:46, Elaine Meng <meng@cgl.ucsf.edu>: >> >> >>>>>>> >> >> >>>>>>> Hello, >> >> >>>>>>> Specifications like "#1.1&protein" already include the >> >> backbone atoms -- they are part of the protein. >> >> >>>>>>> >> >> >>>>>>> >> >>
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
>> >> >>>>>>> >> >> >>>>>>> Maybe the H-bond(s) that you think should be found are less >> >> favorable (longer distance and/or poorer angle). In that case you >> >> could try using larger values than the defaults with the "distSlop" >> >> and/or "angleSlop" options of "hbonds" to make detection more >> >> permissive. >> >> >>>>>>> >> >> >>>>>>> >> >>
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#options>
>> >> >>>>>>> >> >> >>>>>>> I hope this helps, >> >> >>>>>>> Elaine >> >> >>>>>>> ----- >> >> >>>>>>> Elaine C. Meng, Ph.D. >> >> >>>>>>> UCSF Chimera(X) team >> >> >>>>>>> Department of Pharmaceutical Chemistry >> >> >>>>>>> University of California, San Francisco >> >> >>>>>>> >> >> >>>>>>>> On Mar 10, 2022, at 3:28 AM, Enrico Martinez via >> >> ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >> >> >>>>>>>> >> >> >>>>>>>> Dear ChimeraX users! >> >> >>>>>>>> I am using the following command to calculate and save >> in the log >> >> >>>>>>>> information regarding hydrogen bonds based on the >> consideration of >> >> >>>>>>>> multi-frame pdb of the complex >> >> >>>>>>>> >> >> >>>>>>>> # calculate hydrogen bonds for the first 14 frames of pdb >> >> >>>>>>>> hbonds #1.1-14}&protein restrict #1.1-14}&ligand coordsets false >> >> >>>>>>>> interModel false makePseudobonds false log true intraRes false >> >> >>>>>>>> saveFile log.txt >> >> >>>>>>>> >> >> >>>>>>>> which gives me something like this: >> >> >>>>>>>> structure_name: >> >> >>>>>>>> 18 H-bonds >> >> >>>>>>>> H-bonds (donor, acceptor, hydrogen, D..A dist, D-H..A dist): >> >> >>>>>>>> #1.1/? ASN 142 ND2 #1.1/A UNL 1 N #1.1/? ASN 142 >> >> 2HD2 3.313 2.522 >> >> >>>>>>>> #1.1/? SER 144 N #1.1/A UNL 1 O #1.1/? SER 144 >> >> H 2.953 2.224 >> >> >>>>>>>> #1.1/A UNL 1 O #1.1/? LEU 141 O #1.1/A UNL 1 H >> >> 2.753 1.877 >> >> >>>>>>>> #1.2/? ASN 142 ND2 #1.2/A UNL 1 N #1.2/? ASN 142 >> >> 2HD2 3.240 2.429 >> >> >>>>>>>> #1.2/A UNL 1 O #1.2/? HIS 163 NE2 #1.2/A UNL 1 H >> >> 3.317 2.389 >> >> >>>>>>>> #1.3/? SER 144 N #1.3/A UNL 1 O #1.3/? SER 144 >> >> H 3.005 2.272 >> >> >>>>>>>> #1.3/A UNL 1 O #1.3/? LEU 141 O #1.3/A UNL 1 H >> >> 2.738 2.098 >> >> >>>>>>>> #1.3/A UNL 1 O #1.3/? SER 144 OG #1.3/A UNL 1 H >> >> 3.054 2.250 >> >> >>>>>>>> #1.6/A UNL 1 O #1.6/? HIS 163 NE2 #1.6/A UNL 1 H >> >> 3.172 2.528 >> >> >>>>>>>> #1.7/? CYS 145 SG #1.7/A UNL 1 O #1.7/? CYS 145 >> >> HG 3.828 2.949 >> >> >>>>>>>> #1.7/A UNL 1 O #1.7/? LEU 141 O #1.7/A UNL 1 H >> >> 3.201 2.393 >> >> >>>>>>>> #1.8/? GLY 143 N #1.8/A UNL 1 O #1.8/? GLY 143 >> >> H 3.060 2.238 >> >> >>>>>>>> #1.8/? HIS 163 NE2 #1.8/A UNL 1 O no hydrogen >> >> 3.084 N/A >> >> >>>>>>>> #1.9/? GLN 189 NE2 #1.9/A UNL 1 O #1.9/? GLN 189 >> >> 1HE2 3.148 2.139 >> >> >>>>>>>> #1.10/? GLN 189 NE2 #1.10/A UNL 1 O #1.10/? GLN >> >> 189 1HE2 2.941 2.289 >> >> >>>>>>>> #1.10/A UNL 1 O #1.10/? GLN 189 OE1 #1.10/A UNL 1 >> >> H 2.985 2.213 >> >> >>>>>>>> #1.11/? GLN 189 NE2 #1.11/A UNL 1 O #1.11/? GLN >> >> 189 1HE2 2.803 1.910 >> >> >>>>>>>> #1.14/? ASN 142 ND2 #1.14/A UNL 1 O #1.14/? ASN >> >> 142 2HD2 3.199 2.369 >> >> >>>>>>>> >> >> >>
>> >> >>>>>>>> >> >> >>>>>>>> I noticed that in the log there is always information regarding >> >> >>>>>>>> interactions between the ligand and the side chains of the >> >> protein but >> >> >>>>>>>> nothing regarding hydrogen bonds involved backbone of >> the protein >> >> >>>>>>>> (Which is confirmed by X-ray data for my complex). for the >> >> test I used >> >> >>>>>>>> py@ol to visualise h-bonds and may see in the 12th frame >> >> the hydrogen >> >> >>>>>>>> bond involved backbone of the protein.. How could I modify >> >> my script >> >> >>>>>>>> to consider additional hydrogen bonds involving backbone atoms ? >> >> >>>>>>>> Thank you very much in advance! >> >> >>>>>>>> Cheers >> >> >>>>>>>> Enrico >> >> >>>>>>> >> >> >>>>> >> >> >>>> >> >> >>>> >> >> >>>> _______________________________________________ >> >> >>>> ChimeraX-users mailing list >> >> >>>> ChimeraX-users@cgl.ucsf.edu >> >> >>>> Manage subscription: >> >> >>>> https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >> >> >>>> >> >> >>> >> >> >> <test_hbonds-min.png> >> >> > >> >> > >> >> > _______________________________________________ >> >> > ChimeraX-users mailing list >> >> > ChimeraX-users@cgl.ucsf.edu >> >> > Manage subscription: >> >> > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >> >> > >> >> >> > >> > _______________________________________________ >> > ChimeraX-users mailing list >> > ChimeraX-users@cgl.ucsf.edu >> > Manage subscription: >> > https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users >> >> >> >> _______________________________________________ >> ChimeraX-users mailing list >> ChimeraX-users@cgl.ucsf.edu >> Manage subscription: >> https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
participants (5)
-
 Elaine Meng
Elaine Meng -
 Enrico Martinez
Enrico Martinez -
 Guido Hansen
Guido Hansen -
 JAMES MICHAEL S1JJRUdFUiA=
JAMES MICHAEL S1JJRUdFUiA= -
 Tristan Croll
Tristan Croll