Predicting and modeling post-translationally modified C-terminal endings

Hello, I am trying to predict and model a peptide docking to a receptor protein using ChimeraX's AlphaFold interface, and I was wondering if there is any way to specify post-translational modifications in the AlphaFold input function in ChimeraX? Our peptide has an amidated C-terminus, and my searches online have not yielded much success. Thank you, Ryssa Parks

Hi Ryssa, The ChimeraX alphafold tool runs ColabFold, which is based on AlphaFold 2 and does not predict post-translational modifications. <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html> There is another ChimeraX tool that runs Boltz, which is somewhat based on AlphaFold 3. Although the Boltz program can predict post-translational modifications, however, it is a current limitation of the ChimeraX interface (tool and command) that there is no way to specify such modifications. Sorry about that. See <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html#limitations> I hope this clarifies the current situation, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 16, 2025, at 11:16 AM, Parks, Ryssa via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, I am trying to predict and model a peptide docking to a receptor protein using ChimeraX's AlphaFold interface, and I was wondering if there is any way to specify post-translational modifications in the AlphaFold input function in ChimeraX? Our peptide has an amidated C-terminus, and my searches online have not yielded much success.
Thank you, Ryssa Parks

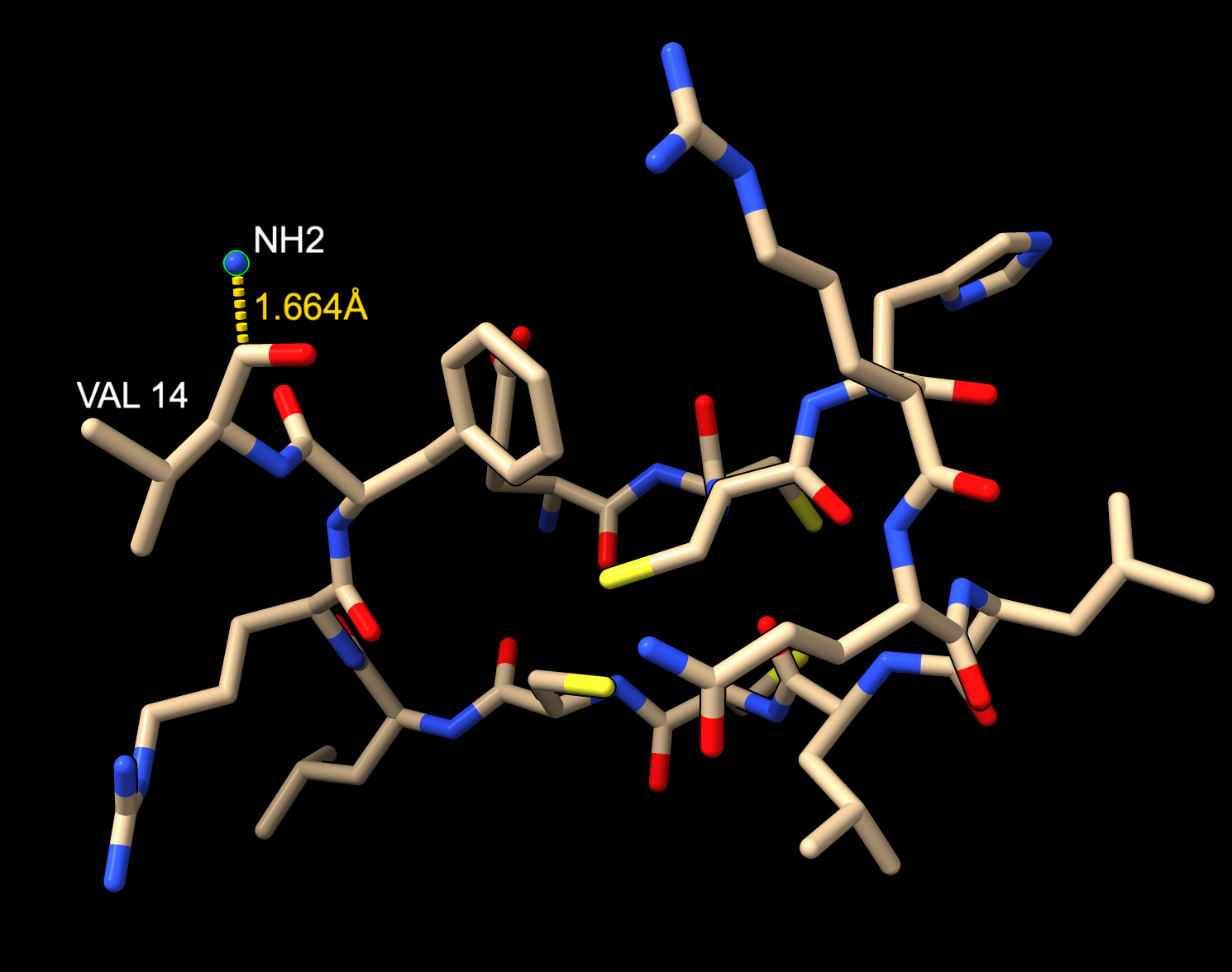
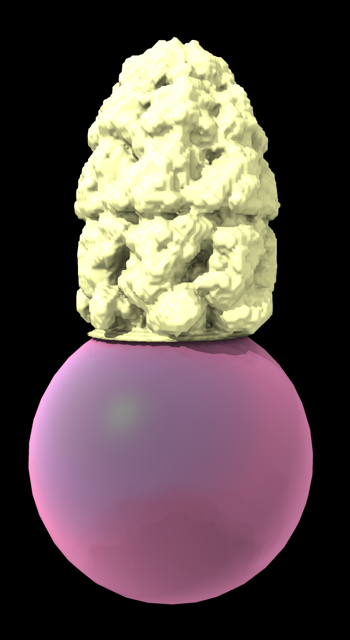
Hi Ryssa, It is possible to use Boltz with ChimeraX to predict an amidated peptide in complex with a protein. As Elaine pointed out the ChimeraX user interface doesn't have a way to do the amidation. But you can run the ChimeraX Boltz tool without the amidation (menu Tools / Structure Prediction / Boltz), and then edit the Boltz input file ChimeraX created to add the amidation and rerun Boltz from a terminal with that new input file. I tried it amidating a 14 amino acid peptide with a valine at the C-terminus. Unfortunately Boltz did not make a good prediction. The added amide was not shown as bonded to the valine, probably that is just a problem with the mmCIF output file, but more troubling the distance to the added amide was too long (1.7 Angstroms), image below. I don't think this is going to be a promising solution for you. But for completeness here are details of what I tried. I used the sequence from amidated-petide structure PDB 3zkt, ECCHRQLLCCLRFV. ChimeraX ran a prediction of this using the following Boltz input file that it created version: 1 sequences: - protein: id: [A] sequence: ECCHRQLLCCLRFV I copied that Boltz input file 3kzt.yaml to a new file 3kzt_nh2.yaml and added the amidation as follows (make sure to use spaces not tabs). version: 1 sequences: - protein: id: [A] sequence: ECCHRQLLCCLRFV - ligand: id: [B] ccd: NH2 constraints: - bond: atom1: [A, 14, C] atom2: [B, 1, N] The amide is added as an NH2 ligand, chain B, and then is connected by a bond to the peptide chain A residue 14, C atom. Then I ran a Boltz prediction with this file from a terminal on my Mac using a modified copy of the command ChimeraX used (found in the file ChimeraX created named "command") $ /Users/goddard/boltz22/bin/boltz predict /Users/goddard/Desktop/boltz/boltz_3zkt/3zkt_nh2.yaml --use_msa_server --accelerator gpu --no_kernels --use_potentials >& output_nh2 I added the option --use_potentials which uses Boltz steering potentials to improve geometry. Without that the amide was 2A away. With steering it got a bit closer at 1.7 A. But the 3zkt PDB has it at 1.3 A. It took about 1 minute. I looked at the Boltz prediction by opening this mmCIF file in ChimeraX ~/Desktop/boltz/boltz_3zkt/boltz_results_3zkt_nh2/predictions/3zkt_nh2/3zkt_nh2_model_0.cif and that is shown below. Tom 14 amino acid peptide with amidated C-terminus predicted by Boltz.
On Sep 16, 2025, at 11:49 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Ryssa, The ChimeraX alphafold tool runs ColabFold, which is based on AlphaFold 2 and does not predict post-translational modifications. <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html>
There is another ChimeraX tool that runs Boltz, which is somewhat based on AlphaFold 3. Although the Boltz program can predict post-translational modifications, however, it is a current limitation of the ChimeraX interface (tool and command) that there is no way to specify such modifications. Sorry about that. See <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html#limitations>
I hope this clarifies the current situation, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 16, 2025, at 11:16 AM, Parks, Ryssa via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, I am trying to predict and model a peptide docking to a receptor protein using ChimeraX's AlphaFold interface, and I was wondering if there is any way to specify post-translational modifications in the AlphaFold input function in ChimeraX? Our peptide has an amidated C-terminus, and my searches online have not yielded much success.
Thank you, Ryssa Parks
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}

Hi Elaine, I am not sure if this appeared already earlier in the BB, but is there a way to compute the inner lumen volume of a particle, which has some large openings (≈40 Å), with ChimeraX? Maybe with some kind of segmentation? Thanks a lot in advance, Xavier On 16/9/25 20:49, Elaine Meng via ChimeraX-users wrote:
Hi Ryssa, The ChimeraX alphafold tool runs ColabFold, which is based on AlphaFold 2 and does not predict post-translational modifications. <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html>
There is another ChimeraX tool that runs Boltz, which is somewhat based on AlphaFold 3. Although the Boltz program can predict post-translational modifications, however, it is a current limitation of the ChimeraX interface (tool and command) that there is no way to specify such modifications. Sorry about that. See <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html#limitations>
I hope this clarifies the current situation, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 16, 2025, at 11:16 AM, Parks, Ryssa via ChimeraX-users<chimerax-users@cgl.ucsf.edu> wrote:
Hello, I am trying to predict and model a peptide docking to a receptor protein using ChimeraX's AlphaFold interface, and I was wondering if there is any way to specify post-translational modifications in the AlphaFold input function in ChimeraX? Our peptide has an amidated C-terminus, and my searches online have not yielded much success.
Thank you, Ryssa Parks
_______________________________________________ ChimeraX-users mailing list --chimerax-users@cgl.ucsf.edu To unsubscribe send an email tochimerax-users-leave@cgl.ucsf.edu Archives:https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
--
{kind=link}
hi Xavier, Better to use a new descriptive subject line rather than extending an unrelated conversation. There is a "Measure Volume and Area" tool (or "measure volume" command) to report volume enclosed in a surface, which could be an isosurface: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/measurevolume.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#volume> However, I don't recall if it handles holes. You could try it. If not, maybe you would need to use some method of generating a surface without holes. As you said, maybe "Segment Map" could do that: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/segment.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 19, 2025, at 8:27 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine, I am not sure if this appeared already earlier in the BB, but is there a way to compute the inner lumen volume of a particle, which has some large openings (≈40 Å), with ChimeraX? Maybe with some kind of segmentation? Thanks a lot in advance, Xavier
Thanks a lot, Elaine. I'll have a try and let you know. On 19/11/25 18:07, Elaine Meng wrote:
hi Xavier, Better to use a new descriptive subject line rather than extending an unrelated conversation.
There is a "Measure Volume and Area" tool (or "measure volume" command) to report volume enclosed in a surface, which could be an isosurface:
<https://rbvi.ucsf.edu/chimerax/docs/user/tools/measurevolume.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#volume>
However, I don't recall if it handles holes. You could try it. If not, maybe you would need to use some method of generating a surface without holes. As you said, maybe "Segment Map" could do that: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/segment.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 19, 2025, at 8:27 AM, F.Xavier Gomis-Rüth via ChimeraX-users<chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine, I am not sure if this appeared already earlier in the BB, but is there a way to compute the inner lumen volume of a particle, which has some large openings (≈40 Å), with ChimeraX? Maybe with some kind of segmentation? Thanks a lot in advance, Xavier
--
{kind=link}
Here is an example of one way to compute an interior cavity volume, in this case a virus capsid, when there are holes. It's a bit tricky. https://rbvi.github.io/chimerax-recipes/virus_volume/virusvol.html Tom
On Nov 19, 2025, at 10:43 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks a lot, Elaine. I'll have a try and let you know.
On 19/11/25 18:07, Elaine Meng wrote:
hi Xavier, Better to use a new descriptive subject line rather than extending an unrelated conversation.
There is a "Measure Volume and Area" tool (or "measure volume" command) to report volume enclosed in a surface, which could be an isosurface:
<https://rbvi.ucsf.edu/chimerax/docs/user/tools/measurevolume.html> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/measurevolume.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#volume> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#volume>
However, I don't recall if it handles holes. You could try it. If not, maybe you would need to use some method of generating a surface without holes. As you said, maybe "Segment Map" could do that: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/segment.html> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/segment.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 19, 2025, at 8:27 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> <mailto:chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine, I am not sure if this appeared already earlier in the BB, but is there a way to compute the inner lumen volume of a particle, which has some large openings (≈40 Å), with ChimeraX? Maybe with some kind of segmentation? Thanks a lot in advance, Xavier
-- <fxgr_signanew.jpg> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Thanks a lot, Tom. The issue is that I have a very wide tunnel connecting the inner and outer surfaces that cannot be erased by a low-resolution map. Is there an option to somehow generate a perpendicular plane or clip the map, "closing" this wide pore? On 19/11/25 20:29, Tom Goddard wrote:
Here is an example of one way to compute an interior cavity volume, in this case a virus capsid, when there are holes. It's a bit tricky.
https://rbvi.github.io/chimerax-recipes/virus_volume/virusvol.html
Tom
On Nov 19, 2025, at 10:43 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks a lot, Elaine. I'll have a try and let you know.
On 19/11/25 18:07, Elaine Meng wrote:
hi Xavier, Better to use a new descriptive subject line rather than extending an unrelated conversation.
There is a "Measure Volume and Area" tool (or "measure volume" command) to report volume enclosed in a surface, which could be an isosurface:
<https://rbvi.ucsf.edu/chimerax/docs/user/tools/measurevolume.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#volume>
However, I don't recall if it handles holes. You could try it. If not, maybe you would need to use some method of generating a surface without holes. As you said, maybe "Segment Map" could do that: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/segment.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 19, 2025, at 8:27 AM, F.Xavier Gomis-Rüth via ChimeraX-users<chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine, I am not sure if this appeared already earlier in the BB, but is there a way to compute the inner lumen volume of a particle, which has some large openings (≈40 Å), with ChimeraX? Maybe with some kind of segmentation? Thanks a lot in advance, Xavier
-- <fxgr_signanew.jpg> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
--
{kind=link}
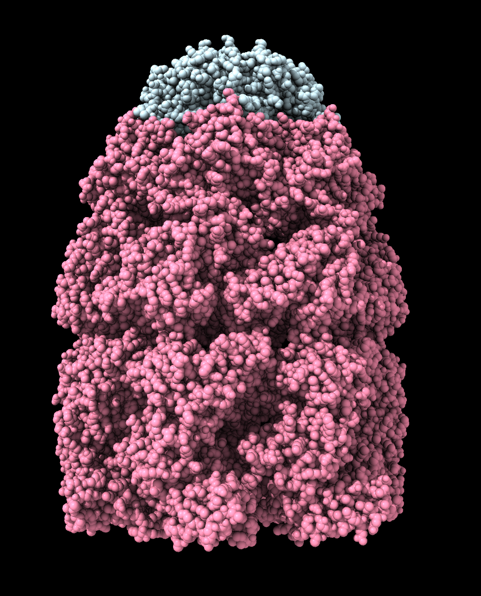
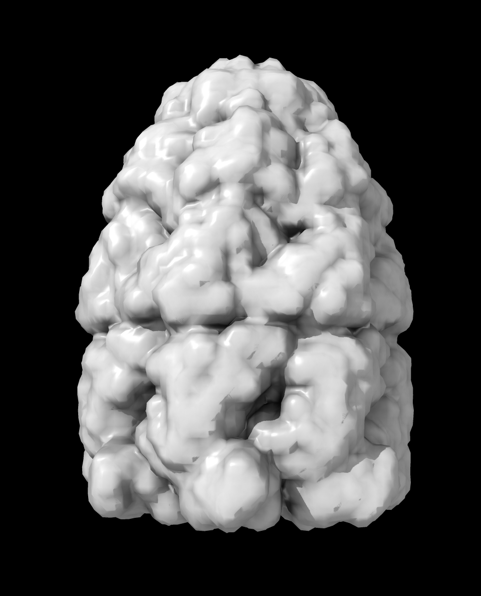
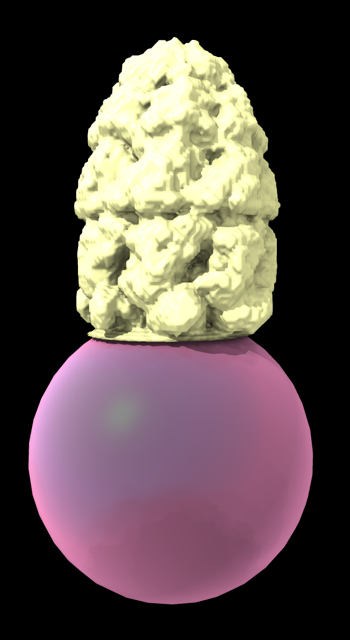
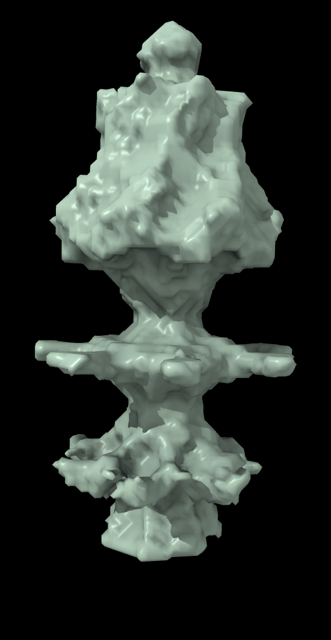
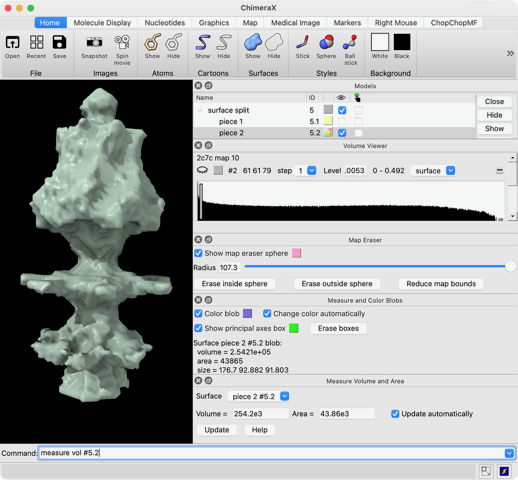
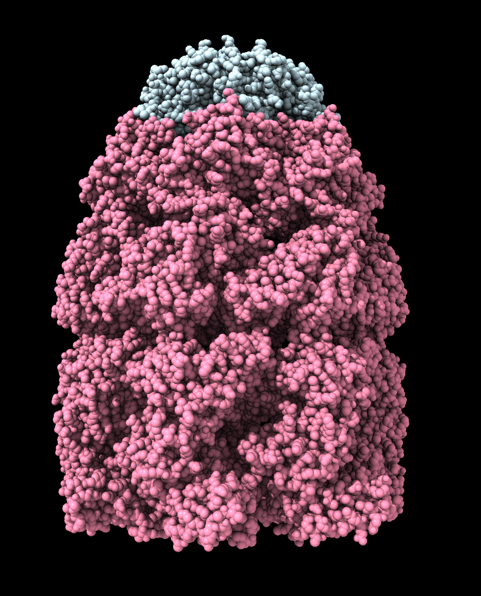
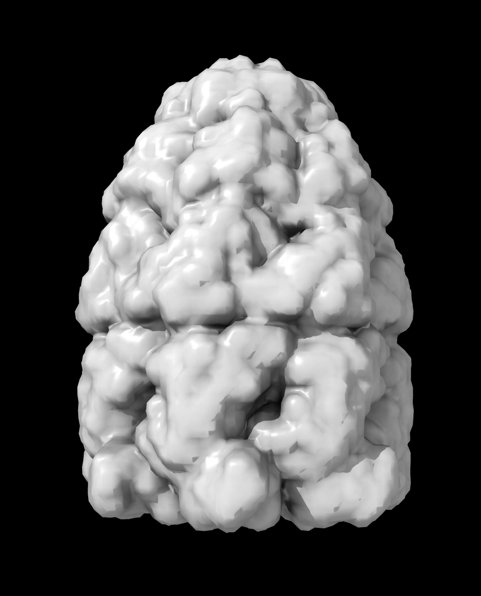
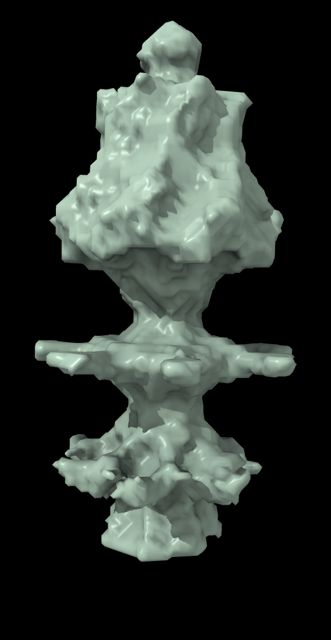
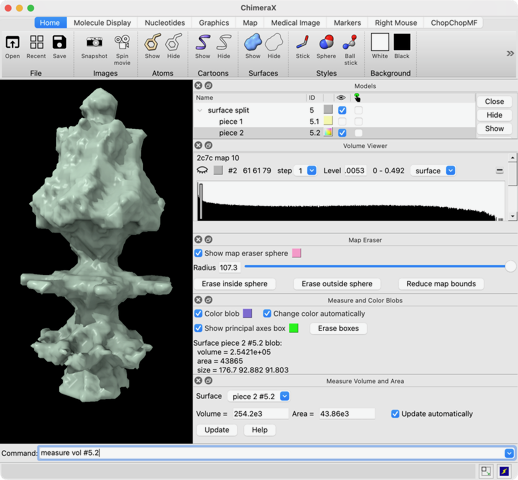
Hi Xavier. ChimeraX can measure the enclosed volume of a surface and put caps over holes when measuring the volume (menu Tools / Volume Data / Measure Volume and Area). But the trick is how do you get such a surface lining just the tunnel. Here's another approach. Make a density map that is 0 inside atoms of a molecular assembly and is 1 outside the assembly. So the 1's region includes the tunnel. But it is connected to the outside region around the molecular assembly via the hole at the end of the tunnel. If we can erase that connection at the hole then we can make a surface of the density in the tunnel region that can be measured. Here's an example using GroES, pdb 2c7c. open 2c7c molmap #1 10 volume onesmask #2 invert true volume #3 cap false # Now use volume eraser, menu Tools / Volume Data / Volume Eraser, make the erasing sphere huge just outside # the tunnel touching the tunnel hole and erase within the sphere. Now the tunnel space density is isolated. # Use Blob mouse mode to color the tunnel density surface (Right Mouse toolbar, Blobs). surface splitbycolor #3 measure volume #5.2 > Enclosed volume for piece 2 (#5.2) = 2.542e+05 I get 254,000 cubic Angstroms for the cavity of GroES. Tom 
On Nov 20, 2025, at 12:32 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks a lot, Tom. The issue is that I have a very wide tunnel connecting the inner and outer surfaces that cannot be erased by a low-resolution map. Is there an option to somehow generate a perpendicular plane or clip the map, "closing" this wide pore?
On 19/11/25 20:29, Tom Goddard wrote:
Here is an example of one way to compute an interior cavity volume, in this case a virus capsid, when there are holes. It's a bit tricky.
https://rbvi.github.io/chimerax-recipes/virus_volume/virusvol.html
Tom
On Nov 19, 2025, at 10:43 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> <mailto:chimerax-users@cgl.ucsf.edu> wrote:
Thanks a lot, Elaine. I'll have a try and let you know.
On 19/11/25 18:07, Elaine Meng wrote:
hi Xavier, Better to use a new descriptive subject line rather than extending an unrelated conversation.
There is a "Measure Volume and Area" tool (or "measure volume" command) to report volume enclosed in a surface, which could be an isosurface:
<https://rbvi.ucsf.edu/chimerax/docs/user/tools/measurevolume.html> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/measurevolume.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#volume> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#volume>
However, I don't recall if it handles holes. You could try it. If not, maybe you would need to use some method of generating a surface without holes. As you said, maybe "Segment Map" could do that: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/segment.html> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/segment.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 19, 2025, at 8:27 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> <mailto:chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine, I am not sure if this appeared already earlier in the BB, but is there a way to compute the inner lumen volume of a particle, which has some large openings (≈40 Å), with ChimeraX? Maybe with some kind of segmentation? Thanks a lot in advance, Xavier
-- <fxgr_signanew.jpg> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
-- <fxgr_signanew.jpg> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Thanks a lot, Tom for you feedback! It worked! On 21/11/25 02:06, Tom Goddard wrote:
Hi Xavier.
ChimeraX can measure the enclosed volume of a surface and put caps over holes when measuring the volume (menu Tools / Volume Data / Measure Volume and Area). But the trick is how do you get such a surface lining just the tunnel.
Here's another approach. Make a density map that is 0 inside atoms of a molecular assembly and is 1 outside the assembly. So the 1's region includes the tunnel. But it is connected to the outside region around the molecular assembly via the hole at the end of the tunnel. If we can erase that connection at the hole then we can make a surface of the density in the tunnel region that can be measured.
Here's an example using GroES, pdb 2c7c.
open 2c7c molmap #1 10 volume onesmask #2 invert true volume #3 cap false # Now use volume eraser, menu Tools / Volume Data / Volume Eraser, make the erasing sphere huge just outside # the tunnel touching the tunnel hole and erase within the sphere. Now the tunnel space density is isolated. # Use Blob mouse mode to color the tunnel density surface (Right Mouse toolbar, Blobs). surface splitbycolor #3 measure volume #5.2 > Enclosed volume for piece 2 (#5.2) = 2.542e+05
I get 254,000 cubic Angstroms for the cavity of GroES.
Tom
2c7c.png2c7c_map.pngmask.pngmask_no_cap.pngerase.pngcolor_blob.pngcavity.pngcavity_volume.png
On Nov 20, 2025, at 12:32 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks a lot, Tom. The issue is that I have a very wide tunnel connecting the inner and outer surfaces that cannot be erased by a low-resolution map. Is there an option to somehow generate a perpendicular plane or clip the map, "closing" this wide pore?
On 19/11/25 20:29, Tom Goddard wrote:
Here is an example of one way to compute an interior cavity volume, in this case a virus capsid, when there are holes. It's a bit tricky.
https://rbvi.github.io/chimerax-recipes/virus_volume/virusvol.html
Tom
On Nov 19, 2025, at 10:43 AM, F.Xavier Gomis-Rüth via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks a lot, Elaine. I'll have a try and let you know.
On 19/11/25 18:07, Elaine Meng wrote:
hi Xavier, Better to use a new descriptive subject line rather than extending an unrelated conversation.
There is a "Measure Volume and Area" tool (or "measure volume" command) to report volume enclosed in a surface, which could be an isosurface:
<https://rbvi.ucsf.edu/chimerax/docs/user/tools/measurevolume.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#volume>
However, I don't recall if it handles holes. You could try it. If not, maybe you would need to use some method of generating a surface without holes. As you said, maybe "Segment Map" could do that: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/segment.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 19, 2025, at 8:27 AM, F.Xavier Gomis-Rüth via ChimeraX-users<chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine, I am not sure if this appeared already earlier in the BB, but is there a way to compute the inner lumen volume of a particle, which has some large openings (≈40 Å), with ChimeraX? Maybe with some kind of segmentation? Thanks a lot in advance, Xavier
-- <fxgr_signanew.jpg> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
-- <fxgr_signanew.jpg> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
--
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
participants (4)
-
 Elaine Meng
Elaine Meng -
 F.Xavier Gomis-Rüth
F.Xavier Gomis-Rüth -
 Parks, Ryssa
Parks, Ryssa -
 Tom Goddard
Tom Goddard