Inquiry About Favorable Contacts and Display Methods in ChimeraX

Subject: Inquiry About Favorable Contacts and Display Methods in ChimeraX Dear ChimeraX Developers, My name is Kenshiro Tabata. I hope this email finds you well. I am currently using ChimeraX to analyze the effects of point mutations on protein structure, particularly in cases where a larger side chain (e.g., methionine) is replaced by a smaller one (e.g., valine). In such cases, I expect that interactions with the opposing amino acids may be reduced due to the decreased side-chain volume. To investigate this, I would like to ask for your guidance regarding the interpretation and visualization of favorable contacts. ________________________________ 1) Understanding Favorable Contacts I understand that the contacts command identifies all types of interactions, whereas the clashes command detects only unfavorable interactions. Based on this, I interpreted the positive VDW overlap as indicating unfavorable (repulsive) interactions, while an overlap between 0 and -0.4 represents favorable (attractive) interactions. Could you confirm if this understanding is correct? ________________________________ 2) Best Approach for Displaying Only Favorable Contacts To analyze how the mutation affects interactions, I would like to display only favorable contacts. Below is the approach I have considered: Proposed Approach 1. Retrieve all interactions (favorable, neutral, and unfavorable) using the contacts command contacts overlapCutoff -0.4 saveFile all_contacts.txt 2. Retrieve only neutral and unfavorable interactions contacts overlapCutoff 0 saveFile non_favorable_contacts.txt 3. Compute the difference between these two sets using Python, extract only favorable contacts, and format them into a ChimeraX pseudobond file # File paths all_contacts_path = "all_contacts.txt" non_favorable_contacts_path = "non_favorable_contacts.txt" favorable_contacts_path = "favorable_contacts.pb" # Read files with open(all_contacts_path, "r") as f: all_contacts = set(f.readlines()) with open(non_favorable_contacts_path, "r") as f: non_favorable_contacts = set(f.readlines()) # Extract favorable contacts favorable_contacts = all_contacts - non_favorable_contacts # Convert to ChimeraX pseudobond file format with open(favorable_contacts_path, "w") as f: f.write("; Pseudobond file for favorable contacts\n") f.write("; radius = 0.1\n; color = green\n; dashes = 6\n") for line in favorable_contacts: parts = line.strip().split() if len(parts) >= 4: atom1 = f"/{parts[0]}:{parts[1]}@{parts[2]}" atom2 = f"/{parts[3]}:{parts[4]}@{parts[5]}" f.write(f"{atom1} {atom2}\n") 4. Load the extracted favorable contacts into ChimeraX for visualization open favorable_contacts.pb Would this be an appropriate approach? Alternatively, is there a simpler or more precise way to achieve this in ChimeraX? I appreciate your time and assistance, and I look forward to your advice. Best regards, Kenshiro Tabata

Dear Kenshiro Tabata, I see a couple of issues with your approach: (1) you are not considering the electrostatic characteristics of the polar interactions. Even if there is a good contact distance between two atoms, they may be electrostatically repulsive and thus unfavorable if both are negatively charged or both are positively charged. So you may want to run "hbonds" to get favorable polar interactions, which are primarily H-bonding. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html> (2) it is not just the VDW overlap cutoff that is different for the "contacts" and "clashes" calculations, but also the "hbAllowance" which is default 0.4 for clashes. This is to allow H-bonding atom pairs to come closer than otherwise expected because of their favorable H-bonding, without being counted as a clash. Your understanding is basically correct, except for this small modification. The "hbAllowance" is explained here: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/clashes.html#hbondAllowanc...> It may not be necessary to run both "contacts" and "clashes" (which are the same except for those two distance-related parameters). Instead we suggest running only a contacts calculation and then filtering the out the clashing results by the overlap value. The overlap cutoff value of 0 in your command is rather restrictive, you may want to allow a small positive to allow for flexibility, but that is your choice. However, since (1) above already includes the favorable polar results, you may want to restrict the contacts calculation to only nonpolar atoms. It is a judgment call how you want to define nonpolar atoms, but some possibilities are: - carbon atoms only, e.g. specified in command line as C - alpha-carbons + sidechains of nonpolar amino acids (another judment call as to which ones you include), for example specified in command line as (sidechain & :ala,val,leu,ile,phe,trp) <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> If you only include nonpolar atoms in the contacts calculation the hbAllowance parameter is irrelevant. So your command could be something like contacts C overlapCutoff -0.4 saveFile all_contacts.txt - OR - contacts (sidechain & :ala,val,leu,ile,phe,trp) overlapCutoff -0.4 saveFile all_contacts.txt However, if you were still planning to include all kinds of atoms in the contacts calculation, including polar, you should probably run with hbAllowance 0.4, e.g. contacts overlapCutoff -0.4 hbAllowance 0.4 saveFile all_contacts.txt After you run the contacts calculation, then you'd edit all_contacts.txt and deletel the rows that have overlap greater than what you want to consider favorable before reformatting into a pb file. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 6, 2025, at 9:31 AM, 田畑 健士郎 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Subject: Inquiry About Favorable Contacts and Display Methods in ChimeraX Dear ChimeraX Developers, My name is Kenshiro Tabata. I hope this email finds you well. I am currently using ChimeraX to analyze the effects of point mutations on protein structure, particularly in cases where a larger side chain (e.g., methionine) is replaced by a smaller one (e.g., valine). In such cases, I expect that interactions with the opposing amino acids may be reduced due to the decreased side-chain volume. To investigate this, I would like to ask for your guidance regarding the interpretation and visualization of favorable contacts.1) Understanding Favorable Contacts I understand that the contacts command identifies all types of interactions, whereas the clashes command detects only unfavorable interactions. Based on this, I interpreted the positive VDW overlap as indicating unfavorable (repulsive) interactions, while an overlap between 0 and -0.4 represents favorable (attractive) interactions. Could you confirm if this understanding is correct?2) Best Approach for Displaying Only Favorable Contacts To analyze how the mutation affects interactions, I would like to display only favorable contacts. Below is the approach I have considered: Proposed Approach • Retrieve all interactions (favorable, neutral, and unfavorable) using the contacts commandcontacts overlapCutoff -0.4 saveFile all_contacts.txt
• Retrieve only neutral and unfavorable interactionscontacts overlapCutoff 0 saveFile non_favorable_contacts.txt
• Compute the difference between these two sets using Python, extract only favorable contacts, and format them into a ChimeraX pseudobond file# File paths all_contacts_path = "all_contacts.txt" non_favorable_contacts_path = "non_favorable_contacts.txt" favorable_contacts_path = "favorable_contacts.pb"
# Read files with open(all_contacts_path, "r") as f: all_contacts = set(f.readlines())
with open(non_favorable_contacts_path, "r") as f: non_favorable_contacts = set(f.readlines())
# Extract favorable contacts favorable_contacts = all_contacts - non_favorable_contacts
# Convert to ChimeraX pseudobond file format with open(favorable_contacts_path, "w") as f: f.write("; Pseudobond file for favorable contacts\n") f.write("; radius = 0.1\n; color = green\n; dashes = 6\n") for line in favorable_contacts: parts = line.strip().split() if len(parts) >= 4: atom1 = f"/{parts[0]}:{parts[1]}@{parts[2]}" atom2 = f"/{parts[3]}:{parts[4]}@{parts[5]}" f.write(f"{atom1} {atom2}\n")
• Load the extracted favorable contacts into ChimeraX for visualizationopen favorable_contacts.pb
Would this be an appropriate approach? Alternatively, is there a simpler or more precise way to achieve this in ChimeraX? I appreciate your time and assistance, and I look forward to your advice. Best regards, Kenshiro Tabata _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Dear Elaine, I sincerely appreciate your detailed and thoughtful explanation. Your guidance has been incredibly helpful, and I now have a much clearer understanding of the best approach. Based on your advice, here is the workflow I plan to follow: 1) Use the hbonds command to extract hydrogen bonds. 2) Use contacts (sidechain & :ala,val,leu,ile,phe,trp) overlapCutoff -0.4 saveFile all_contacts.txt to extract interactions among nonpolar atoms. 3) Remove entries from all_contacts.txt where the overlap is 0 or a small positive value, to exclude unfavorable interactions. 4) Combine the results from steps (1) and (3), convert them into .pb format, and visualize them in ChimeraX. Alternatively, they can be displayed separately with different colors for clarity. In step 2, when focusing on a specific pair of amino acids, I noticed that multiple pseudobonds were drawn between atoms, connecting them (I have attached an image as an example). In this case, one amino acid is methionine, and the other is tryptophan. Would it be correct to interpret that these multiple pseudobonds collectively represent a hydrophobic interaction? If there are any other refinements or insights you would recommend, I would be very grateful to hear them. Once again, I truly appreciate your time and support. Your kind and detailed advice has been invaluable, and I’m very thankful for your help. Best regards, Kenshiro Tabata ________________________________ 差出人: Elaine Meng <meng@cgl.ucsf.edu> 送信日時: 2025年3月7日 7:05 宛先: 田畑 健士郎 <tabata-k@ncnp.go.jp> CC: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> 件名: Re: [chimerax-users] Inquiry About Favorable Contacts and Display Methods in ChimeraX Dear Kenshiro Tabata, I see a couple of issues with your approach: (1) you are not considering the electrostatic characteristics of the polar interactions. Even if there is a good contact distance between two atoms, they may be electrostatically repulsive and thus unfavorable if both are negatively charged or both are positively charged. So you may want to run "hbonds" to get favorable polar interactions, which are primarily H-bonding. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html> (2) it is not just the VDW overlap cutoff that is different for the "contacts" and "clashes" calculations, but also the "hbAllowance" which is default 0.4 for clashes. This is to allow H-bonding atom pairs to come closer than otherwise expected because of their favorable H-bonding, without being counted as a clash. Your understanding is basically correct, except for this small modification. The "hbAllowance" is explained here: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/clashes.html#hbondAllowanc...> It may not be necessary to run both "contacts" and "clashes" (which are the same except for those two distance-related parameters). Instead we suggest running only a contacts calculation and then filtering the out the clashing results by the overlap value. The overlap cutoff value of 0 in your command is rather restrictive, you may want to allow a small positive to allow for flexibility, but that is your choice. However, since (1) above already includes the favorable polar results, you may want to restrict the contacts calculation to only nonpolar atoms. It is a judgment call how you want to define nonpolar atoms, but some possibilities are: - carbon atoms only, e.g. specified in command line as C - alpha-carbons + sidechains of nonpolar amino acids (another judment call as to which ones you include), for example specified in command line as (sidechain & :ala,val,leu,ile,phe,trp) <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin> If you only include nonpolar atoms in the contacts calculation the hbAllowance parameter is irrelevant. So your command could be something like contacts C overlapCutoff -0.4 saveFile all_contacts.txt - OR - contacts (sidechain & :ala,val,leu,ile,phe,trp) overlapCutoff -0.4 saveFile all_contacts.txt However, if you were still planning to include all kinds of atoms in the contacts calculation, including polar, you should probably run with hbAllowance 0.4, e.g. contacts overlapCutoff -0.4 hbAllowance 0.4 saveFile all_contacts.txt After you run the contacts calculation, then you'd edit all_contacts.txt and deletel the rows that have overlap greater than what you want to consider favorable before reformatting into a pb file. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 6, 2025, at 9:31 AM, 田畑 健士郎 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Subject: Inquiry About Favorable Contacts and Display Methods in ChimeraX Dear ChimeraX Developers, My name is Kenshiro Tabata. I hope this email finds you well. I am currently using ChimeraX to analyze the effects of point mutations on protein structure, particularly in cases where a larger side chain (e.g., methionine) is replaced by a smaller one (e.g., valine). In such cases, I expect that interactions with the opposing amino acids may be reduced due to the decreased side-chain volume. To investigate this, I would like to ask for your guidance regarding the interpretation and visualization of favorable contacts.1) Understanding Favorable Contacts I understand that the contacts command identifies all types of interactions, whereas the clashes command detects only unfavorable interactions. Based on this, I interpreted the positive VDW overlap as indicating unfavorable (repulsive) interactions, while an overlap between 0 and -0.4 represents favorable (attractive) interactions. Could you confirm if this understanding is correct?2) Best Approach for Displaying Only Favorable Contacts To analyze how the mutation affects interactions, I would like to display only favorable contacts. Below is the approach I have considered: Proposed Approach • Retrieve all interactions (favorable, neutral, and unfavorable) using the contacts commandcontacts overlapCutoff -0.4 saveFile all_contacts.txt
• Retrieve only neutral and unfavorable interactionscontacts overlapCutoff 0 saveFile non_favorable_contacts.txt
• Compute the difference between these two sets using Python, extract only favorable contacts, and format them into a ChimeraX pseudobond file# File paths all_contacts_path = "all_contacts.txt" non_favorable_contacts_path = "non_favorable_contacts.txt" favorable_contacts_path = "favorable_contacts.pb"
# Read files with open(all_contacts_path, "r") as f: all_contacts = set(f.readlines())
with open(non_favorable_contacts_path, "r") as f: non_favorable_contacts = set(f.readlines())
# Extract favorable contacts favorable_contacts = all_contacts - non_favorable_contacts
# Convert to ChimeraX pseudobond file format with open(favorable_contacts_path, "w") as f: f.write("; Pseudobond file for favorable contacts\n") f.write("; radius = 0.1\n; color = green\n; dashes = 6\n") for line in favorable_contacts: parts = line.strip().split() if len(parts) >= 4: atom1 = f"/{parts[0]}:{parts[1]}@{parts[2]}" atom2 = f"/{parts[3]}:{parts[4]}@{parts[5]}" f.write(f"{atom1} {atom2}\n")
• Load the extracted favorable contacts into ChimeraX for visualizationopen favorable_contacts.pb
Would this be an appropriate approach? Alternatively, is there a simpler or more precise way to achieve this in ChimeraX? I appreciate your time and assistance, and I look forward to your advice. Best regards, Kenshiro Tabata _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
Hi Kenshiro, You have to use your own chemical intuition, whether you think that interaction is hydrophobic or not. Those residues also have some polar character so it probably depends who you ask on whether it is a polar interaction or a hydrophobic interaction, or both. Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 7, 2025, at 5:01 AM, 田畑 健士郎 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Elaine,
I sincerely appreciate your detailed and thoughtful explanation. Your guidance has been incredibly helpful, and I now have a much clearer understanding of the best approach.
Based on your advice, here is the workflow I plan to follow:
1) Use the hbonds command to extract hydrogen bonds. 2) Use contacts (sidechain & :ala,val,leu,ile,phe,trp) overlapCutoff -0.4 saveFile all_contacts.txt to extract interactions among nonpolar atoms. 3) Remove entries from all_contacts.txt where the overlap is 0 or a small positive value, to exclude unfavorable interactions. 4) Combine the results from steps (1) and (3), convert them into .pb format, and visualize them in ChimeraX. Alternatively, they can be displayed separately with different colors for clarity.
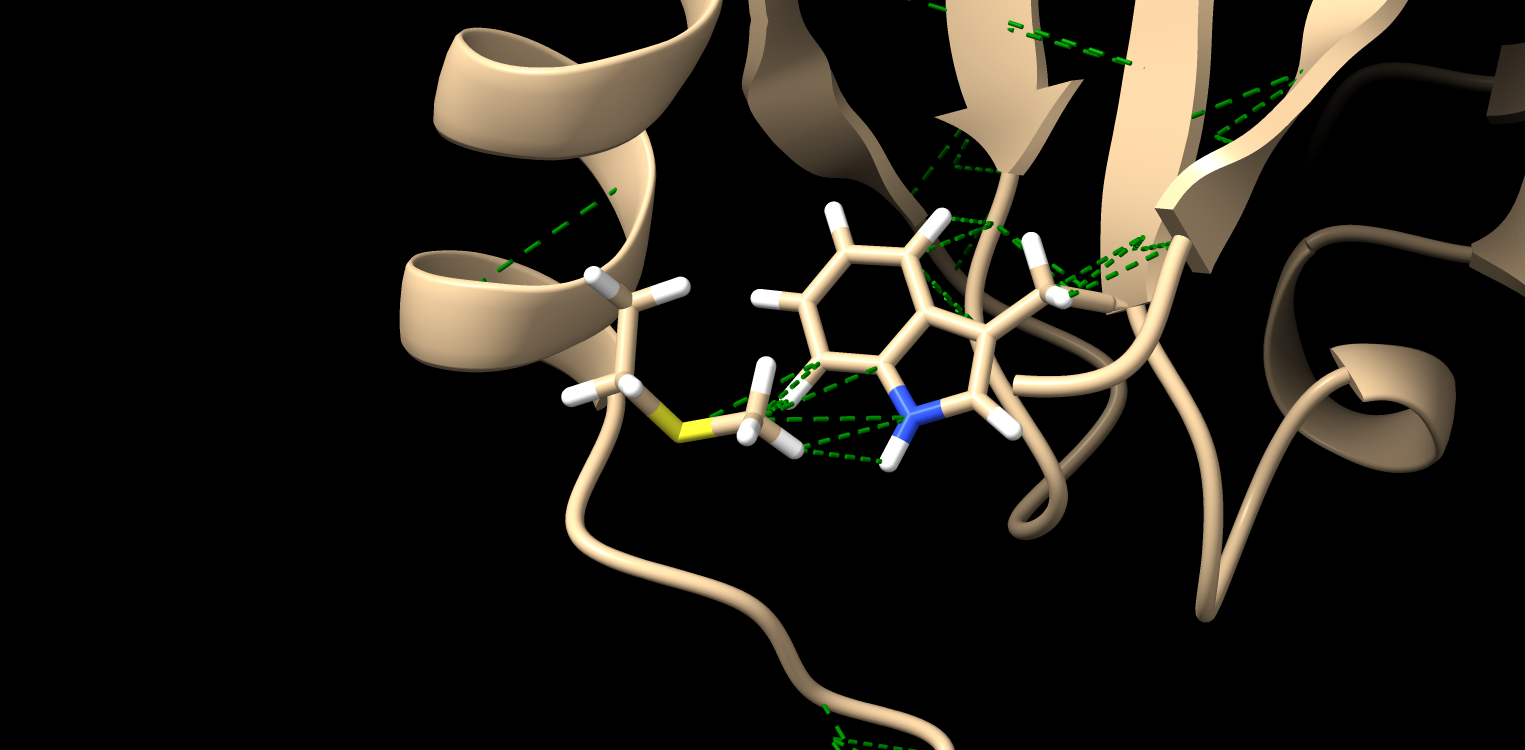
In step 2, when focusing on a specific pair of amino acids, I noticed that multiple pseudobonds were drawn between atoms, connecting them (I have attached an image as an example). In this case, one amino acid is methionine, and the other is tryptophan. Would it be correct to interpret that these multiple pseudobonds collectively represent a hydrophobic interaction?
If there are any other refinements or insights you would recommend, I would be very grateful to hear them.
Once again, I truly appreciate your time and support. Your kind and detailed advice has been invaluable, and I’m very thankful for your help.
Best regards, Kenshiro Tabata 差出人: Elaine Meng <meng@cgl.ucsf.edu> 送信日時: 2025年3月7日 7:05 宛先: 田畑 健士郎 <tabata-k@ncnp.go.jp> CC: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> 件名: Re: [chimerax-users] Inquiry About Favorable Contacts and Display Methods in ChimeraX Dear Kenshiro Tabata, I see a couple of issues with your approach:
(1) you are not considering the electrostatic characteristics of the polar interactions. Even if there is a good contact distance between two atoms, they may be electrostatically repulsive and thus unfavorable if both are negatively charged or both are positively charged. So you may want to run "hbonds" to get favorable polar interactions, which are primarily H-bonding. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html>
(2) it is not just the VDW overlap cutoff that is different for the "contacts" and "clashes" calculations, but also the "hbAllowance" which is default 0.4 for clashes. This is to allow H-bonding atom pairs to come closer than otherwise expected because of their favorable H-bonding, without being counted as a clash. Your understanding is basically correct, except for this small modification. The "hbAllowance" is explained here: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/clashes.html#hbondAllowanc...>
It may not be necessary to run both "contacts" and "clashes" (which are the same except for those two distance-related parameters). Instead we suggest running only a contacts calculation and then filtering the out the clashing results by the overlap value. The overlap cutoff value of 0 in your command is rather restrictive, you may want to allow a small positive to allow for flexibility, but that is your choice.
However, since (1) above already includes the favorable polar results, you may want to restrict the contacts calculation to only nonpolar atoms. It is a judgment call how you want to define nonpolar atoms, but some possibilities are: - carbon atoms only, e.g. specified in command line as C - alpha-carbons + sidechains of nonpolar amino acids (another judment call as to which ones you include), for example specified in command line as (sidechain & :ala,val,leu,ile,phe,trp) <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin>
If you only include nonpolar atoms in the contacts calculation the hbAllowance parameter is irrelevant. So your command could be something like
contacts C overlapCutoff -0.4 saveFile all_contacts.txt - OR - contacts (sidechain & :ala,val,leu,ile,phe,trp) overlapCutoff -0.4 saveFile all_contacts.txt
However, if you were still planning to include all kinds of atoms in the contacts calculation, including polar, you should probably run with hbAllowance 0.4, e.g.
contacts overlapCutoff -0.4 hbAllowance 0.4 saveFile all_contacts.txt
After you run the contacts calculation, then you'd edit all_contacts.txt and deletel the rows that have overlap greater than what you want to consider favorable before reformatting into a pb file. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 6, 2025, at 9:31 AM, 田畑 健士郎 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Subject: Inquiry About Favorable Contacts and Display Methods in ChimeraX Dear ChimeraX Developers, My name is Kenshiro Tabata. I hope this email finds you well. I am currently using ChimeraX to analyze the effects of point mutations on protein structure, particularly in cases where a larger side chain (e.g., methionine) is replaced by a smaller one (e.g., valine). In such cases, I expect that interactions with the opposing amino acids may be reduced due to the decreased side-chain volume. To investigate this, I would like to ask for your guidance regarding the interpretation and visualization of favorable contacts.1) Understanding Favorable Contacts I understand that the contacts command identifies all types of interactions, whereas the clashes command detects only unfavorable interactions. Based on this, I interpreted the positive VDW overlap as indicating unfavorable (repulsive) interactions, while an overlap between 0 and -0.4 represents favorable (attractive) interactions. Could you confirm if this understanding is correct?2) Best Approach for Displaying Only Favorable Contacts To analyze how the mutation affects interactions, I would like to display only favorable contacts. Below is the approach I have considered: Proposed Approach • Retrieve all interactions (favorable, neutral, and unfavorable) using the contacts commandcontacts overlapCutoff -0.4 saveFile all_contacts.txt
• Retrieve only neutral and unfavorable interactionscontacts overlapCutoff 0 saveFile non_favorable_contacts.txt
• Compute the difference between these two sets using Python, extract only favorable contacts, and format them into a ChimeraX pseudobond file# File paths all_contacts_path = "all_contacts.txt" non_favorable_contacts_path = "non_favorable_contacts.txt" favorable_contacts_path = "favorable_contacts.pb"
# Read files with open(all_contacts_path, "r") as f: all_contacts = set(f.readlines())
with open(non_favorable_contacts_path, "r") as f: non_favorable_contacts = set(f.readlines())
# Extract favorable contacts favorable_contacts = all_contacts - non_favorable_contacts
# Convert to ChimeraX pseudobond file format with open(favorable_contacts_path, "w") as f: f.write("; Pseudobond file for favorable contacts\n") f.write("; radius = 0.1\n; color = green\n; dashes = 6\n") for line in favorable_contacts: parts = line.strip().split() if len(parts) >= 4: atom1 = f"/{parts[0]}:{parts[1]}@{parts[2]}" atom2 = f"/{parts[3]}:{parts[4]}@{parts[5]}" f.write(f"{atom1} {atom2}\n")
• Load the extracted favorable contacts into ChimeraX for visualizationopen favorable_contacts.pb
Would this be an appropriate approach? Alternatively, is there a simpler or more precise way to achieve this in ChimeraX? I appreciate your time and assistance, and I look forward to your advice. Best regards, Kenshiro Tabata _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
<Met_Trp.png>_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
participants (2)
-
 Elaine Meng
Elaine Meng -
 田畑 健士郎
田畑 健士郎