
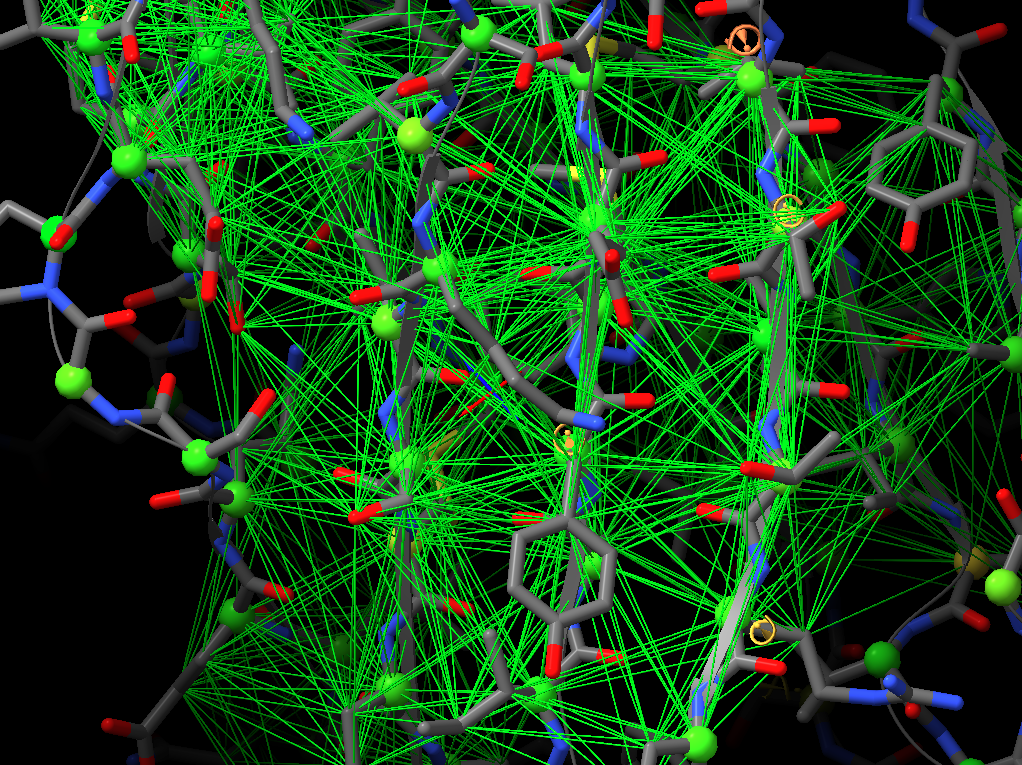
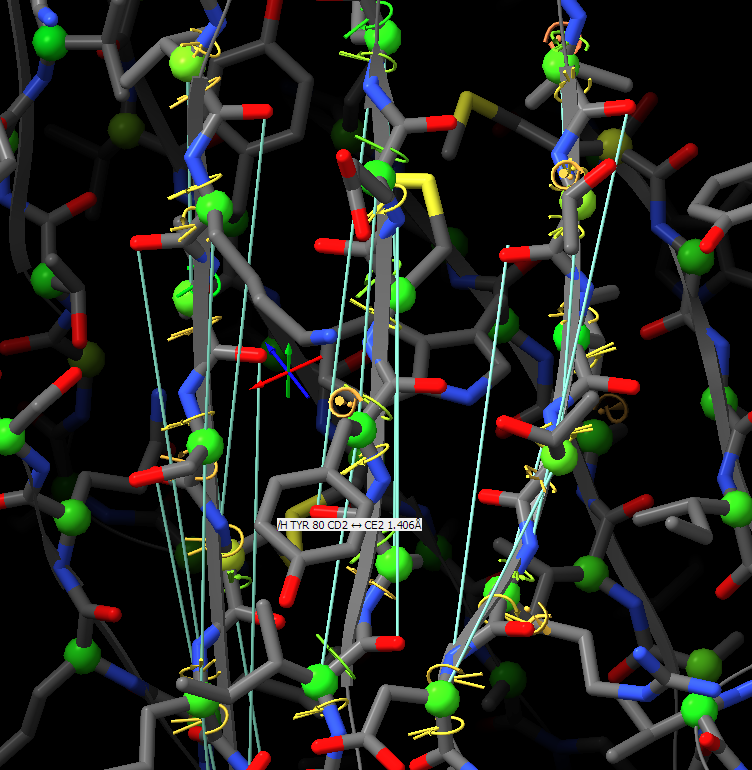
Oops! My bad - was responding on the move, and forgetting my own implementation. You're right, the "isolde restrain distances" command works on a per-residue basis, so sidechain atoms will be included regardless. On the other hand, the "isolde release distances" command works per-atom, so after the restrain command you can selectively release the sidechain restraints with: isolde release distances #1&sideonly ________________________________ From: Guido Hansen <hansen@biochem.uni-luebeck.de> Sent: 15 March 2022 16:20 To: Tristan Croll <tic20@cam.ac.uk> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] ISOLDE and secondary structure restraints Thanks for your suggestions. However, I think that when applying distance restraints using the command below also distances of mainchain atoms of secondary structure elements to nearby sidechain atoms get restrainted. isolde restrain distances #1&(helix|strand)&backbone [cid:part1.09p0uiHA.41fj6uxZ@biochem.uni-luebeck.de] What you get when using the gui is something like that: [cid:part2.0mNP8JTc.JBo6Z60y@biochem.uni-luebeck.de] -- Guido Am 13.03.2022 um 16:07 schrieb Tristan Croll: Not the hard-coded helix and beta-sheet restraints, I’m afraid. But you can get a very similar (probably better, TBH) result with something like: isolde restrain distances #1&(helix|strand)&backbone isolde restrain torsions #1&(helix|strand) sidechain false The advantage of those is that (a) they’ll work a bit better when the true conformation isn’t *perfect* helix or strand, and (b) they have a lot more flexibility in terms of your ability to fine-tune their behaviour (for models fetched from the Alphafold DB, try adding the argument “adjustForConfidence true” to automatically take the pLDDT and PAE values into account). — Tristan On 13 Mar 2022, at 14:42, Guido Hansen via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hi all, I just started model building in cryoEM maps and get great results using ISOLDE on AlphaFold2-derived initial models. I have a question which is mainly directed to Tristan I guess: Is is possible to use the command line/script to apply secondary structure restraints in ISOLDE? I would like to restrain the initial secondary structure assignment from Alphafold especially at resolution worse than 3.5 A. At the moment I select each and every residue range assigned to helix, parallel or antiparallel beta-strand one after the other and turn on the corresponding restrains in the ISOLDE GUI. Is there a better way via command line of script? Cheers, Guido -- PD Dr. Guido Hansen Group Leader [Uni-Lübeck] Universität zu Lübeck Institut für Biochemie Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de<mailto:hansen@biochem.uni-luebeck.de> www.biochem.uni-luebeck.de<http://www.biochem.uni-luebeck.de> Ratzeburger Allee 160 23562 Lübeck _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu<mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users -- PD Dr. Guido Hansen Group Leader [Uni-Lübeck] Universität zu Lübeck Institut für Biochemie Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de<mailto:hansen@biochem.uni-luebeck.de> www.biochem.uni-luebeck.de<http://www.biochem.uni-luebeck.de> Ratzeburger Allee 160 23562 Lübeck
{kind=link}
{kind=link}