
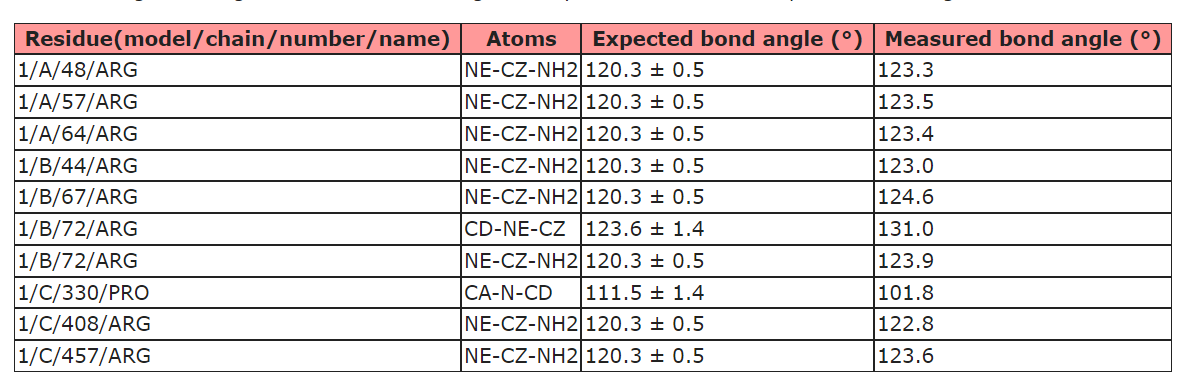
Hi Guido, Yes, there are a few small discrepancies between the AMBER force field and the libraries used for restraints/validation in refinement packages and the wwPDB. That's one of the reasons I try not to describe ISOLDE as a true refinement package (the other being that it currently doesn't perform B-factor refinement). You'll still want to run a final refinement with your package of choice before depositing to the wwPDB. To help with that, there are the commands: isolde write phenixRefineInput isolde write phenixRsrInput isolde write refmacRestraints Just type "usage isolde write" on the command line for more details on their use. Best, Tristan ________________________________ From: ChimeraX-users <chimerax-users-bounces@cgl.ucsf.edu> on behalf of Guido Hansen via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Sent: 16 March 2022 17:38 To: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: [chimerax-users] Bond angles of Arg in ISOLDE It seems that the parameterization of ISOLDE's force field differs from the metrics used by the official PBD validation server as I usually get complaints about Arg bond angles from the validation server (see below) even though everything seems fine in ISOLDE. Is there anything that I can/have to do about that? Or is that something that you would keep as it is and discuss with the PDB people? [cid:11d377ce14f2ac4dcd683be5424ebf5d@biochem.uni-luebeck.de] - Guido
{kind=link}