Can I get atomic coordinates from an electron density map in xplor format via ChimeraX software?

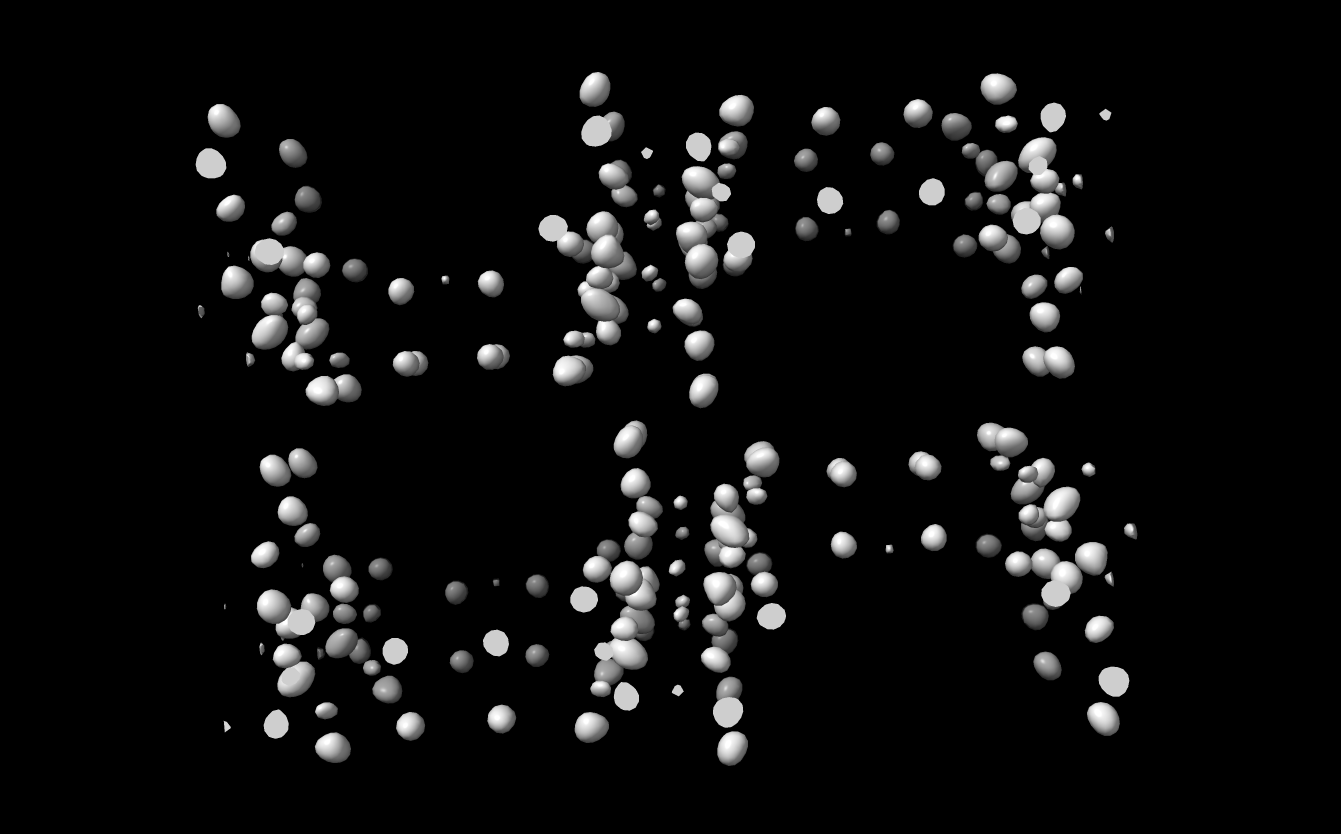{kind=link}

Dear Wang Chao, Sorry no, you cannot automatically save atoms if all you have is a density map. A map can only be saved as as a map, not as atomic coordinates. You would have to build the atomic structure yourself, based on looking at the map. However, as far as I know that is not really feasible with ChimeraX.... There is an ISOLDE plugin for building/refinement into density maps, but I believe it is for organic structures, primarily proteins, not something like a zeolite. <https://cxtoolshed.rbvi.ucsf.edu/apps/chimeraxisolde> One similar thing you could do in ChimeraX, however, is to place "markers" on the density spots, optionally with "links" between the markers, using the Marker Placement tool (menu Tools... Volume Data... Marker Placement). <http://rbvi.ucsf.edu/chimerax/docs/user/tools/markerplacement.html> Markers and links are like dummy atoms and bonds, and they can be saved to marker file, which also remembers the display colors and radii. <http://rbvi.ucsf.edu/chimerax/docs/user/markers.html> You can also save a set of markers and links to an atomic file like PDB or mmCIF (which do not remember the colors and radii). However, this process would not automatically make the bond lengths and angles chemically reasonable, and there is no way to associate these markers and links with the correct elements and their parameters to minimize them to a chemically reasonable structure. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 28, 2020, at 1:23 AM, 王超 <wangchao17@mails.jlu.edu.cn> wrote:
Dear professor I found ChimeraX software from the Internet. Through interface design, I think this should be a very powerful software. Now, I have a electron density map of zeolite in ''.xplor'' format. By adjusting the threshold, I can clearly see where the atom is. So I wonder whether it is possible to use this software to export the coordinates of the structural atoms,especially ''.cif '' format files. Thank you for your help ! -- Best Wishes!

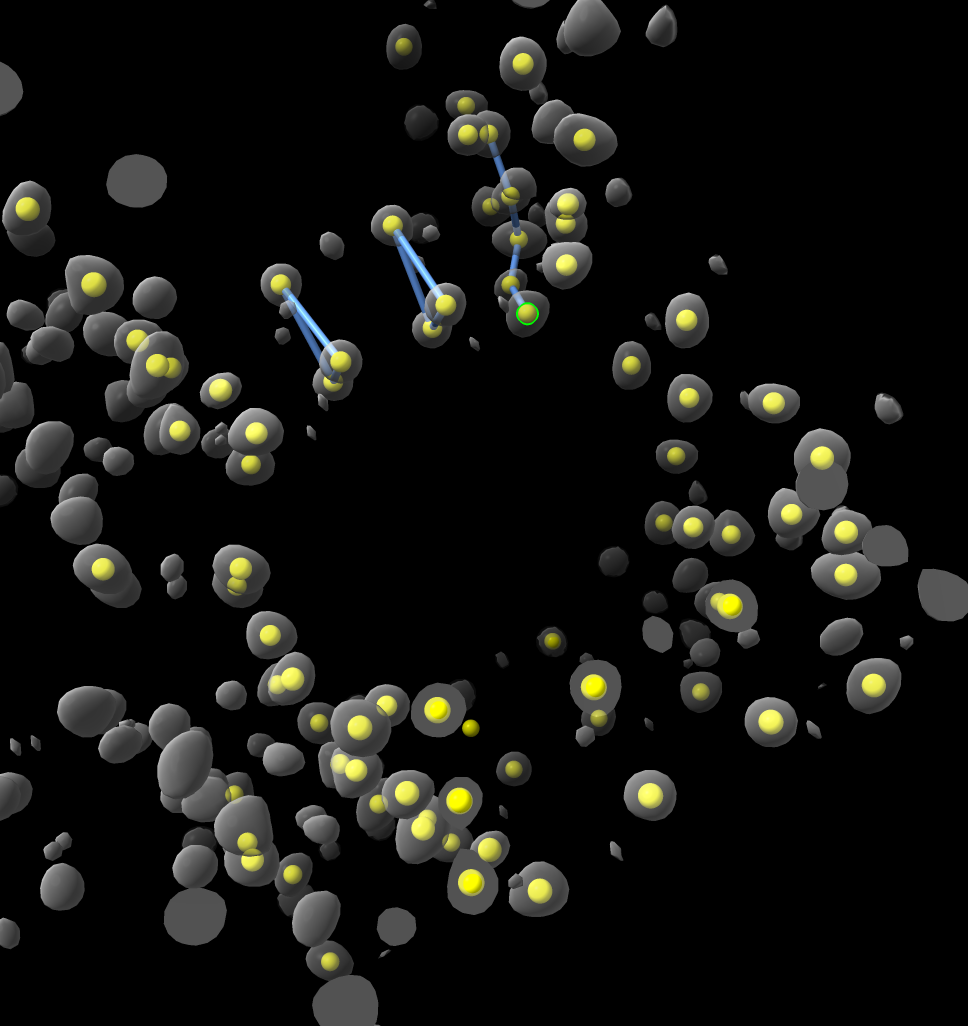
Hi Wang, I think as Elaine suggests just placing markers on the density map might get you started. I tried it using the ChimeraX Markers toolbar using place markers on Center mouse mode. Then just right-clicked 50 blobs to place markers, then used the Link markers mouse mode to connect a few, then wrote out an mmCIF file. All seemed doable. Of course the atom types, silicon vs oxygen are not distinguished but you could perhaps hand edit the mmCIF file to fix that. Tom
On Jul 28, 2020, at 1:23 AM, 王超 <wangchao17@mails.jlu.edu.cn> wrote:
Dear professor I found ChimeraX software from the Internet. Through interface design, I think this should be a very powerful software. Now, I have a electron density map of zeolite in ''.xplor'' format. By adjusting the threshold, I can clearly see where the atom is. So I wonder whether it is possible to use this software to export the coordinates of the structural atoms,especially ''.cif '' format files. Thank you for your help !
-- Best Wishes!
Wang, Chao Jilin University 2699 Qianjin Street, Changchun 130012, China Email:wangchao17@mails.jlu.edu.cn <mailto:wangchao17@mails.jlu.edu.cn> <IFT.png><IFT.xplor>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
participants (3)
-
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard -
 王超
王超