
Hello, My name is Yunsik Kang, and I am a postdoc in Marc Freeman's lab at the Vollum Institute. I would love to use ChimeraX to predict the structure of my protein of interest. I watched all the YouTube videos and tried to run the program. Unfortunately, my protein is 5005 amino acids in humans and 2958 aa in yeast. I get a message "Please use the full AlphaFold system for long sequences." My question is what is the best way to approach this problem? Should I cut the protein in half and run the program? In one of the videos, it mentions after 700 aa it will have problems. Will it work if I get Colab-Pro? Or would the server crash no matter what. I am not a structural biologist, but I hope the structure can help be predict me with my research. Thank you, Yunsik

Hi Yunsik, I don't know where/how you can run a prediction for such a long sequence, maybe others can comment on that. However: First make sure there is not already a predicted structure for that protein in the AlphaFold Database! E.g. try the Search button on the AlphaFold tool and see if the same UniProt entries as you are interested in are found. The database includes human and other well-studied species (maybe also the yeast you mentioned, depending on the exact species). If the prediction is already in the database, there is no need to run a new calculation. ChimeraX AlphaFold tool: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 8, 2021, at 2:45 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
My name is Yunsik Kang, and I am a postdoc in Marc Freeman’s lab at the Vollum Institute.
I would love to use ChimeraX to predict the structure of my protein of interest. I watched all the YouTube videos and tried to run the program.
Unfortunately, my protein is 5005 amino acids in humans and 2958 aa in yeast. I get a message “Please use the full AlphaFold system for long sequences.”
My question is what is the best way to approach this problem? Should I cut the protein in half and run the program? In one of the videos, it mentions after 700 aa it will have problems. Will it work if I get Colab-Pro? Or would the server crash no matter what.
I am not a structural biologist, but I hope the structure can help be predict me with my research.
Thank you, Yunsik

The human protein will be in the EBI-Alphafold2 database, but not accessible to the search tool. For human proteins (and only human, so far) longer than ~2000 residues, the predictions are provided as multiple 1400-residue fragments starting at residue 1, 201, etc.. The only way to get those right now is to download the tarball of the complete set of human protein predictions, and find them in there.
On 8 Sep 2021, at 23:08, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Yunsik, I don't know where/how you can run a prediction for such a long sequence, maybe others can comment on that. However:
First make sure there is not already a predicted structure for that protein in the AlphaFold Database! E.g. try the Search button on the AlphaFold tool and see if the same UniProt entries as you are interested in are found. The database includes human and other well-studied species (maybe also the yeast you mentioned, depending on the exact species). If the prediction is already in the database, there is no need to run a new calculation.
ChimeraX AlphaFold tool: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 8, 2021, at 2:45 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
My name is Yunsik Kang, and I am a postdoc in Marc Freeman’s lab at the Vollum Institute.
I would love to use ChimeraX to predict the structure of my protein of interest. I watched all the YouTube videos and tried to run the program.
Unfortunately, my protein is 5005 amino acids in humans and 2958 aa in yeast. I get a message “Please use the full AlphaFold system for long sequences.”
My question is what is the best way to approach this problem? Should I cut the protein in half and run the program? In one of the videos, it mentions after 700 aa it will have problems. Will it work if I get Colab-Pro? Or would the server crash no matter what.
I am not a structural biologist, but I hope the structure can help be predict me with my research.
Thank you, Yunsik
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users

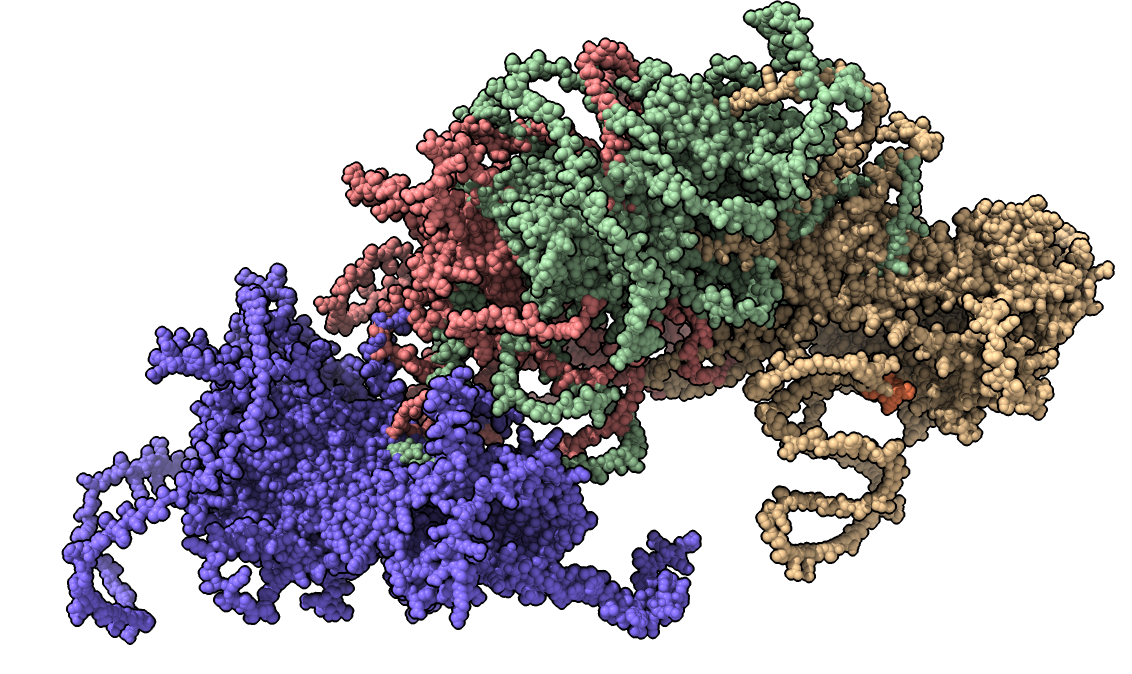
Hi Yunsik, Inspired by your question and Tristan Croll's comment that the AlphaFold database has human proteins longer than 1400 amino acids computed as separate 1400 amino acid chunks I made a ChimeraX command "bigalpha" that loads and aligns these chunks. https://rbvi.github.io/chimerax-recipes/big_alphafold/bigalpha.html I attach two images and a PDB model made by running that command in ChimeraX for 5005 amino acid transmembrane protein Q2LD37 | K1109_HUMAN Transmembrane protein KIAA1109 OS=Homo sapiens OX=9606 GN=KIAA1109 PE=1 SV=2 and also here are a few other examples I looked at yesterday. https://twitter.com/UCSFChimeraX/status/1435870388043411458 <https://twitter.com/UCSFChimeraX/status/1435870388043411458> https://twitter.com/UCSFChimeraX/status/1435760774824169474 <https://twitter.com/UCSFChimeraX/status/1435760774824169474> https://twitter.com/UCSFChimeraX/status/1435859111053193216 <https://twitter.com/UCSFChimeraX/status/1435859111053193216> Keep in mind that the pieces of these models are not aligned in a reliable way and probably clash badly with each other because AlphaFold did not compute them as part of one structure. So these structures should only give you a very rough idea about the protein. Tom This is the confidence coloring determined by AlphaFold, red low, blue high confidence. To reproduce this color using ChimeraX daily build open the PDB model and type command "color bfactor #1 palette alphafold". (The confidence value is saved as the bfactor in the PDB file). Different AlphaFold chunks have different colors and chain identifiers in the PDB model.
On Sep 8, 2021, at 2:45 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
My name is Yunsik Kang, and I am a postdoc in Marc Freeman’s lab at the Vollum Institute.
I would love to use ChimeraX to predict the structure of my protein of interest. I watched all the YouTube videos and tried to run the program.
Unfortunately, my protein is 5005 amino acids in humans and 2958 aa in yeast. I get a message “Please use the full AlphaFold system for long sequences.”
My question is what is the best way to approach this problem? Should I cut the protein in half and run the program? In one of the videos, it mentions after 700 aa it will have problems. Will it work if I get Colab-Pro? Or would the server crash no matter what.
I am not a structural biologist, but I hope the structure can help be predict me with my research.
Thank you, Yunsik
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users <https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users>
{kind=link}
{kind=link}
Hi Tom, Thank you so much! It is great that I inspired you to write this command. Since I have you (and I am no expert in structural biology), do you think alphafold will soon update larger proteins? Is there still a limitation? On your Youtube videos you guys state after 700 aa the system crashes a lot. If I know a protein that perhaps might have a similar structure to my protein would it help resolves these clashing problems? As you can see I tried to hide what my protein of interest was but it seems there are not a lot of 5005 amino acid proteins =). Thank you again! Yunsik From: Tom Goddard <goddard@sonic.net> Sent: Thursday, September 9, 2021 3:17 PM To: Yunsik Kang <kangy@ohsu.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: [EXTERNAL] AlphaFold model for large proteins Hi Yunsik, Inspired by your question and Tristan Croll's comment that the AlphaFold database has human proteins longer than 1400 amino acids computed as separate 1400 amino acid chunks I made a ChimeraX command "bigalpha" that loads and aligns these chunks. https://rbvi.github.io/chimerax-recipes/big_alphafold/bigalpha.html I attach two images and a PDB model made by running that command in ChimeraX for 5005 amino acid transmembrane protein Q2LD37 | K1109_HUMAN Transmembrane protein KIAA1109 OS=Homo sapiens OX=9606 GN=KIAA1109 PE=1 SV=2 and also here are a few other examples I looked at yesterday. https://twitter.com/UCSFChimeraX/status/1435870388043411458 https://twitter.com/UCSFChimeraX/status/1435760774824169474 https://twitter.com/UCSFChimeraX/status/1435859111053193216 Keep in mind that the pieces of these models are not aligned in a reliable way and probably clash badly with each other because AlphaFold did not compute them as part of one structure. So these structures should only give you a very rough idea about the protein. Tom This is the confidence coloring determined by AlphaFold, red low, blue high confidence. To reproduce this color using ChimeraX daily build open the PDB model and type command "color bfactor #1 palette alphafold". (The confidence value is saved as the bfactor in the PDB file). [cid:image001.png@01D7A590.EC640310] Different AlphaFold chunks have different colors and chain identifiers in the PDB model. [cid:image002.png@01D7A590.EC640310] On Sep 8, 2021, at 2:45 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hello, My name is Yunsik Kang, and I am a postdoc in Marc Freeman’s lab at the Vollum Institute. I would love to use ChimeraX to predict the structure of my protein of interest. I watched all the YouTube videos and tried to run the program. Unfortunately, my protein is 5005 amino acids in humans and 2958 aa in yeast. I get a message “Please use the full AlphaFold system for long sequences.” My question is what is the best way to approach this problem? Should I cut the protein in half and run the program? In one of the videos, it mentions after 700 aa it will have problems. Will it work if I get Colab-Pro? Or would the server crash no matter what. I am not a structural biologist, but I hope the structure can help be predict me with my research. Thank you, Yunsik _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu<mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://plato.cgl.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
{kind=link}
Hi Yunsik, I've seen some Twitter messages that say AlphaFold might handle 2000 amino acids if you have say 4 GPUs and allow it to use memory from all of them and possibly do other tricks. But the limitation is that graphics processors have only so much memory and AlphaFold wants a lot, and more for larger sequences. So I don't think it is possible to compute a longer sequence as a single structure currently. It certainly is possible to combine the AlphaFold 1400 amino acid chunks in more sensible ways trying to avoid clashes but I don't have time to pursue that. I figured you might not have wanted to mention your protein, but if you say it has 5005 amino acids then the cat is out of the bag. Tom
On Sep 9, 2021, at 3:39 PM, Yunsik Kang <kangy@ohsu.edu> wrote:
Hi Tom,
Thank you so much! It is great that I inspired you to write this command.
Since I have you (and I am no expert in structural biology), do you think alphafold will soon update larger proteins? Is there still a limitation? On your Youtube videos you guys state after 700 aa the system crashes a lot.
If I know a protein that perhaps might have a similar structure to my protein would it help resolves these clashing problems?
As you can see I tried to hide what my protein of interest was but it seems there are not a lot of 5005 amino acid proteins =).
Thank you again! Yunsik
From: Tom Goddard <goddard@sonic.net <mailto:goddard@sonic.net>> Sent: Thursday, September 9, 2021 3:17 PM To: Yunsik Kang <kangy@ohsu.edu <mailto:kangy@ohsu.edu>> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: [EXTERNAL] AlphaFold model for large proteins
Hi Yunsik,
Inspired by your question and Tristan Croll's comment that the AlphaFold database has human proteins longer than 1400 amino acids computed as separate 1400 amino acid chunks I made a ChimeraX command "bigalpha" that loads and aligns these chunks.
https://rbvi.github.io/chimerax-recipes/big_alphafold/bigalpha.html <https://rbvi.github.io/chimerax-recipes/big_alphafold/bigalpha.html>
I attach two images and a PDB model made by running that command in ChimeraX for 5005 amino acid transmembrane protein
Q2LD37 | K1109_HUMAN Transmembrane protein KIAA1109 OS=Homo sapiens OX=9606 GN=KIAA1109 PE=1 SV=2
and also here are a few other examples I looked at yesterday.
https://twitter.com/UCSFChimeraX/status/1435870388043411458 <https://twitter.com/UCSFChimeraX/status/1435870388043411458>
https://twitter.com/UCSFChimeraX/status/1435760774824169474 <https://twitter.com/UCSFChimeraX/status/1435760774824169474>
https://twitter.com/UCSFChimeraX/status/1435859111053193216 <https://twitter.com/UCSFChimeraX/status/1435859111053193216>
Keep in mind that the pieces of these models are not aligned in a reliable way and probably clash badly with each other because AlphaFold did not compute them as part of one structure. So these structures should only give you a very rough idea about the protein.
Tom
This is the confidence coloring determined by AlphaFold, red low, blue high confidence. To reproduce this color using ChimeraX daily build open the PDB model and type command "color bfactor #1 palette alphafold". (The confidence value is saved as the bfactor in the PDB file).
Different AlphaFold chunks have different colors and chain identifiers in the PDB model.
On Sep 8, 2021, at 2:45 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hello,
My name is Yunsik Kang, and I am a postdoc in Marc Freeman’s lab at the Vollum Institute.
I would love to use ChimeraX to predict the structure of my protein of interest. I watched all the YouTube videos and tried to run the program.
Unfortunately, my protein is 5005 amino acids in humans and 2958 aa in yeast. I get a message “Please use the full AlphaFold system for long sequences.”
My question is what is the best way to approach this problem? Should I cut the protein in half and run the program? In one of the videos, it mentions after 700 aa it will have problems. Will it work if I get Colab-Pro? Or would the server crash no matter what.
I am not a structural biologist, but I hope the structure can help be predict me with my research.
Thank you, Yunsik
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users <https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users>
It's important to remember that the red-to-orange parts have low confidence scores and "should not be interpreted" so it is not meaningful whether or not they are clashing. See: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html#coloring> Elaine
On Sep 9, 2021, at 3:39 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Tom,
Thank you so much! It is great that I inspired you to write this command.
Since I have you (and I am no expert in structural biology), do you think alphafold will soon update larger proteins? Is there still a limitation? On your Youtube videos you guys state after 700 aa the system crashes a lot.
If I know a protein that perhaps might have a similar structure to my protein would it help resolves these clashing problems?
As you can see I tried to hide what my protein of interest was but it seems there are not a lot of 5005 amino acid proteins =).
Thank you again! Yunsik
From: Tom Goddard <goddard@sonic.net> Sent: Thursday, September 9, 2021 3:17 PM To: Yunsik Kang <kangy@ohsu.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: [EXTERNAL] AlphaFold model for large proteins
Hi Yunsik,
Inspired by your question and Tristan Croll's comment that the AlphaFold database has human proteins longer than 1400 amino acids computed as separate 1400 amino acid chunks I made a ChimeraX command "bigalpha" that loads and aligns these chunks.
https://rbvi.github.io/chimerax-recipes/big_alphafold/bigalpha.html
I attach two images and a PDB model made by running that command in ChimeraX for 5005 amino acid transmembrane protein
Q2LD37 | K1109_HUMAN Transmembrane protein KIAA1109 OS=Homo sapiens OX=9606 GN=KIAA1109 PE=1 SV=2
and also here are a few other examples I looked at yesterday.
https://twitter.com/UCSFChimeraX/status/1435870388043411458
https://twitter.com/UCSFChimeraX/status/1435760774824169474
https://twitter.com/UCSFChimeraX/status/1435859111053193216
Keep in mind that the pieces of these models are not aligned in a reliable way and probably clash badly with each other because AlphaFold did not compute them as part of one structure. So these structures should only give you a very rough idea about the protein.
Tom
This is the confidence coloring determined by AlphaFold, red low, blue high confidence. To reproduce this color using ChimeraX daily build open the PDB model and type command "color bfactor #1 palette alphafold". (The confidence value is saved as the bfactor in the PDB file). <image001.png>
Different AlphaFold chunks have different colors and chain identifiers in the PDB model. <image002.png>
On Sep 8, 2021, at 2:45 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
My name is Yunsik Kang, and I am a postdoc in Marc Freeman’s lab at the Vollum Institute.
I would love to use ChimeraX to predict the structure of my protein of interest. I watched all the YouTube videos and tried to run the program.
Unfortunately, my protein is 5005 amino acids in humans and 2958 aa in yeast. I get a message “Please use the full AlphaFold system for long sequences.”
My question is what is the best way to approach this problem? Should I cut the protein in half and run the program? In one of the videos, it mentions after 700 aa it will have problems. Will it work if I get Colab-Pro? Or would the server crash no matter what.
I am not a structural biologist, but I hope the structure can help be predict me with my research.
Thank you, Yunsik
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Thank you Elaine. This really helps. It seems there is no easy I can run the whole protein. Thank you again. This is a great program! Yk -----Original Message----- From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Thursday, September 9, 2021 4:54 PM To: Yunsik Kang <kangy@ohsu.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: [EXTERNAL] Re: [chimerax-users] AlphaFold model for large proteins It's important to remember that the red-to-orange parts have low confidence scores and "should not be interpreted" so it is not meaningful whether or not they are clashing. See: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html#coloring> Elaine
On Sep 9, 2021, at 3:39 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Tom,
Thank you so much! It is great that I inspired you to write this command.
Since I have you (and I am no expert in structural biology), do you think alphafold will soon update larger proteins? Is there still a limitation? On your Youtube videos you guys state after 700 aa the system crashes a lot.
If I know a protein that perhaps might have a similar structure to my protein would it help resolves these clashing problems?
As you can see I tried to hide what my protein of interest was but it seems there are not a lot of 5005 amino acid proteins =).
Thank you again! Yunsik
From: Tom Goddard <goddard@sonic.net> Sent: Thursday, September 9, 2021 3:17 PM To: Yunsik Kang <kangy@ohsu.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: [EXTERNAL] AlphaFold model for large proteins
Hi Yunsik,
Inspired by your question and Tristan Croll's comment that the AlphaFold database has human proteins longer than 1400 amino acids computed as separate 1400 amino acid chunks I made a ChimeraX command "bigalpha" that loads and aligns these chunks.
https://rbvi.github.io/chimerax-recipes/big_alphafold/bigalpha.html
I attach two images and a PDB model made by running that command in ChimeraX for 5005 amino acid transmembrane protein
Q2LD37 | K1109_HUMAN Transmembrane protein KIAA1109 OS=Homo sapiens OX=9606 GN=KIAA1109 PE=1 SV=2
and also here are a few other examples I looked at yesterday.
https://twitter.com/UCSFChimeraX/status/1435870388043411458
https://twitter.com/UCSFChimeraX/status/1435760774824169474
https://twitter.com/UCSFChimeraX/status/1435859111053193216
Keep in mind that the pieces of these models are not aligned in a reliable way and probably clash badly with each other because AlphaFold did not compute them as part of one structure. So these structures should only give you a very rough idea about the protein.
Tom
This is the confidence coloring determined by AlphaFold, red low, blue high confidence. To reproduce this color using ChimeraX daily build open the PDB model and type command "color bfactor #1 palette alphafold". (The confidence value is saved as the bfactor in the PDB file). <image001.png>
Different AlphaFold chunks have different colors and chain identifiers in the PDB model. <image002.png>
On Sep 8, 2021, at 2:45 PM, Yunsik Kang via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
My name is Yunsik Kang, and I am a postdoc in Marc Freeman’s lab at the Vollum Institute.
I would love to use ChimeraX to predict the structure of my protein of interest. I watched all the YouTube videos and tried to run the program.
Unfortunately, my protein is 5005 amino acids in humans and 2958 aa in yeast. I get a message “Please use the full AlphaFold system for long sequences.”
My question is what is the best way to approach this problem? Should I cut the protein in half and run the program? In one of the videos, it mentions after 700 aa it will have problems. Will it work if I get Colab-Pro? Or would the server crash no matter what.
I am not a structural biologist, but I hope the structure can help be predict me with my research.
Thank you, Yunsik
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
participants (4)
-
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard -
 Tristan Croll
Tristan Croll -
 Yunsik Kang
Yunsik Kang