fitmap command in chimerax

Greeting there developer, I am using fitmap command of chimerax to fit pdb to map, the command as below: fitmap #2,3 inMap #1 seq 2 res 3.7 #2 and #3 are copy of each other and I manually placed them in different positions in map, #2 and #3 come to same position after the command running, I had shift option false, but come to same result. I remember it works in chimera. Could you please point me out? Thanks and have a nice day, Yanhe

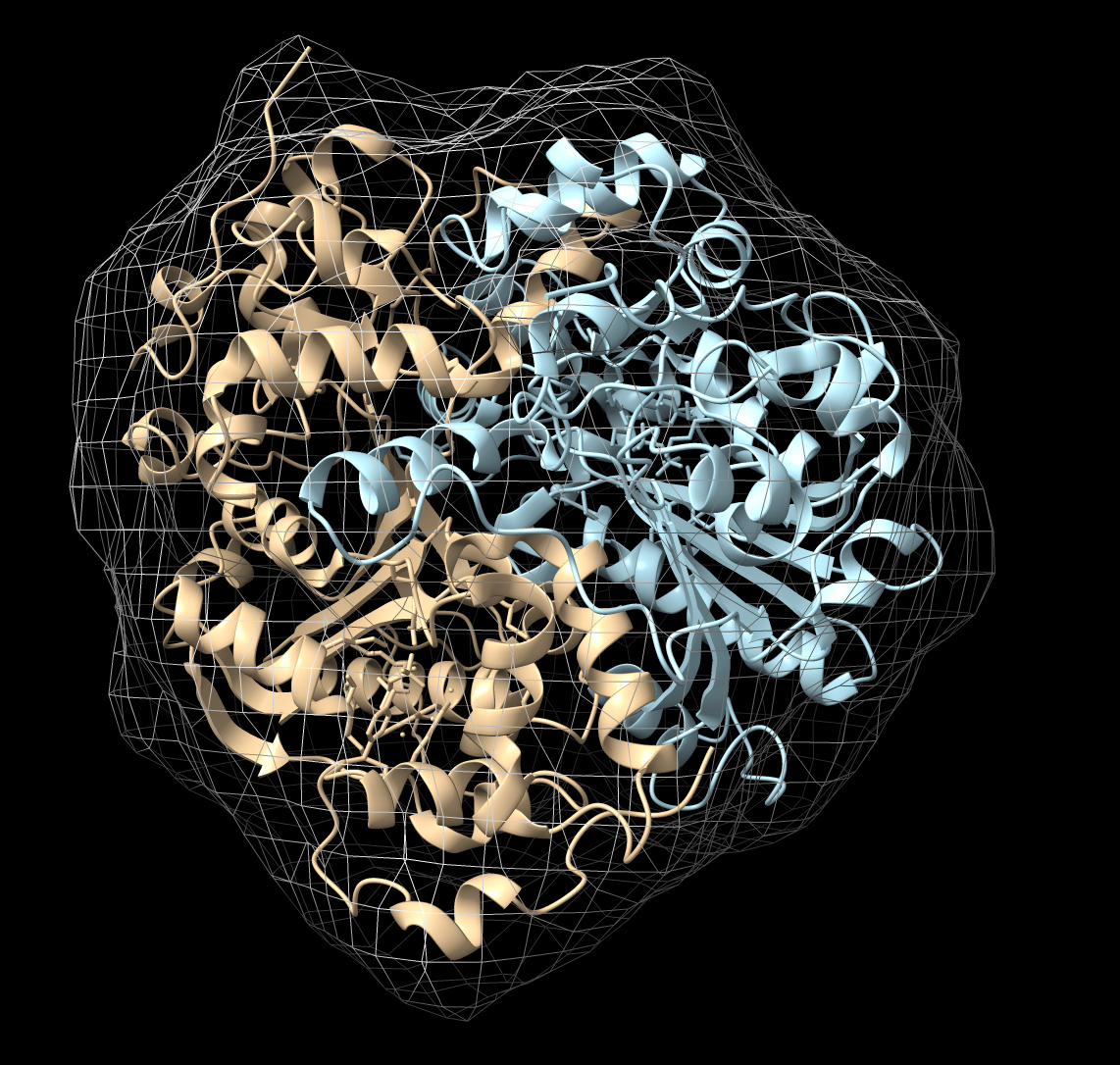
Hi Yanhe, The fit sequential mode which fits each atomic model after subtracting the density occupied by other atomic models appears to be working find in ChimeraX 1.4 in my tests. So I think it has something to do with your map and models and their initial placement. Here is an test example I just tried. I opened a dimer atomic model, made a 10A map from it, split the dimer into two monomer models, move one of the monomers so it collides with the other, then fit the two monomers. open 7ej3 molmap #1 10 split #1 color #1.2 lightblue move x 10 model #1.1 coord #1.1 fit #1.1-2 in #2 seq 2 res 10 It worked as expected pushing the monomers apart so they fit nicely without clashing. To debug what is going wrong in your specific case you can do the sequential fitting steps by hand. Make a simulated map from your second monomer with the molmap command, subtract it from your map using volume subtract with the "minrms true" option so the simulated map is first scaled to match the experimental map, then fit the first monomer in that difference map. Now make a molmap simulated map of the new first monomer position, subtract it from the original experimental map and fit the second monomer into the result. This is exactly the process that the fitmap command with the sequential option is using. Doing it step by step will probably reveal your problem. A simpler approach could be to use "fit #2,3 in #1 seq 1 res 3.7" to see just the first step (fitting only model #2 after subtracting the density occupied by #3), judge whether the result makes sense, then use "fit #3,2 in #1 seq 1 res 3.7" to take one more step fitting only model #3 after subtracting the density occupied by #2. Tom Sequential fitting resulting from example commands on 7ej3
On Sep 21, 2022, at 8:17 AM, Yanhe Zhao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Greeting there developer,
I am using fitmap command of chimerax to fit pdb to map, the command as below: fitmap #2,3 inMap #1 seq 2 res 3.7 #2 and #3 are copy of each other and I manually placed them in different positions in map, #2 and #3 come to same position after the command running, I had shift option false, but come to same result. I remember it works in chimera. Could you please point me out?
Thanks and have a nice day, Yanhe
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}

Hi Yanhe, I could not reproduce this problem. Even if I have the two copies of the same atomic model in exactly the same place before I run the command: fitmap #2,3 inMap #1 seq 2 res 3.7 ... they go to different places. But if you use "shift false" it will prevent shifting during fitting, so I don't think you want to use that option. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 21, 2022, at 8:17 AM, Yanhe Zhao via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Greeting there developer,
I am using fitmap command of chimerax to fit pdb to map, the command as below: fitmap #2,3 inMap #1 seq 2 res 3.7 #2 and #3 are copy of each other and I manually placed them in different positions in map, #2 and #3 come to same position after the command running, I had shift option false, but come to same result. I remember it works in chimera. Could you please point me out?
Thanks and have a nice day, Yanhe
Dear Tom and Elaine, Thanks a lot for your favorable reply. I will most likely follow Tom's idea: do seq fit manually. Thanks and have a nice day, Yanhe On Wed, Sep 21, 2022, 1:07 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Yanhe, I could not reproduce this problem. Even if I have the two copies of the same atomic model in exactly the same place before I run the command:
fitmap #2,3 inMap #1 seq 2 res 3.7
... they go to different places. But if you use "shift false" it will prevent shifting during fitting, so I don't think you want to use that option. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 21, 2022, at 8:17 AM, Yanhe Zhao via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Greeting there developer,
I am using fitmap command of chimerax to fit pdb to map, the command as below: fitmap #2,3 inMap #1 seq 2 res 3.7 #2 and #3 are copy of each other and I manually placed them in different positions in map, #2 and #3 come to same position after the command running, I had shift option false, but come to same result. I remember it works in chimera. Could you please point me out?
Thanks and have a nice day, Yanhe
participants (3)
-
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard -
 Yanhe Zhao
Yanhe Zhao