Does ChimeraX incorporate bond geometry or torsional state into atom display?

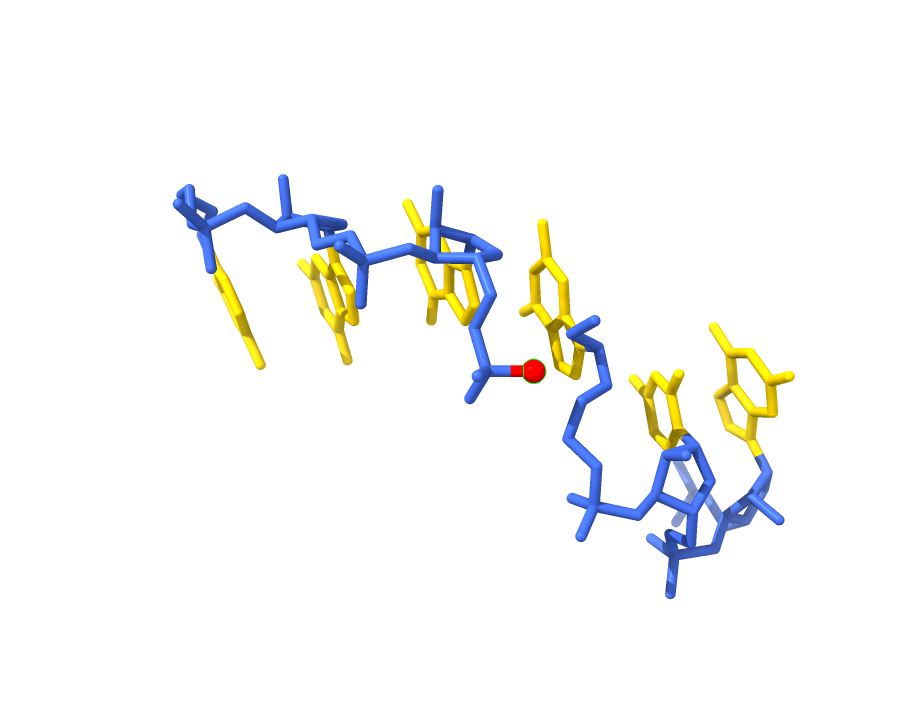
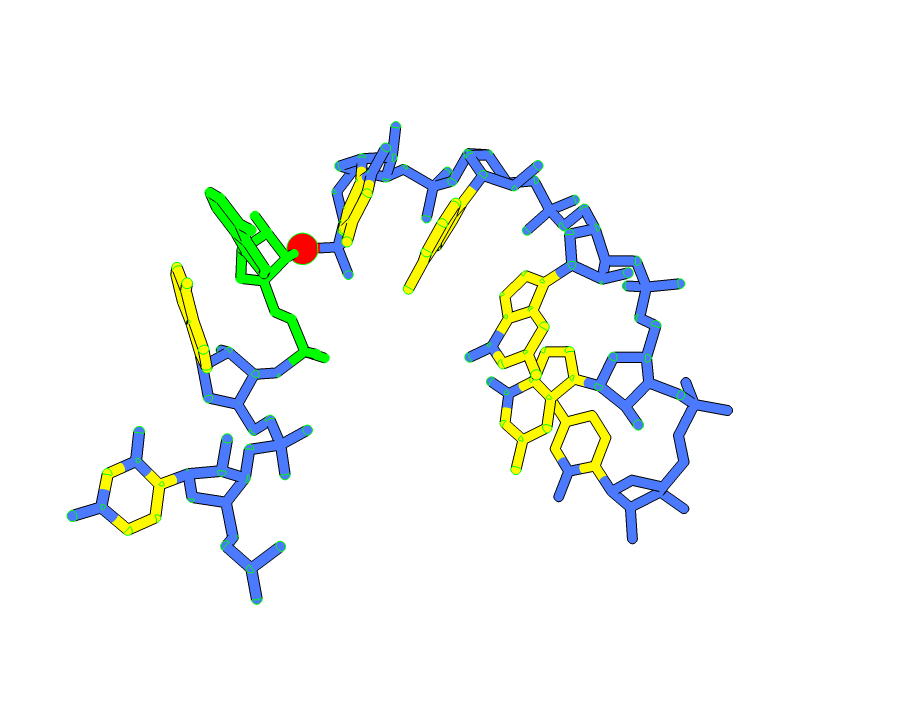
Hi, My nameis Wes Dennis and I am a retired Geophysicist who is just interested in stuff. I am trying to understand what I am seeing with two ChimeraX images. I applied identical atom-name based coloring commands to the same residue range in two different crystal structures of the same molecule — backbone atoms colored blue, aromatic base atoms colored yellow. The visual result is strikingly different between the two structures. In one, blue color appears to spread into the base panels — a structure in which torsional stresses are present. In the other, where torsional stresses are absent, the base panels are clean yellow with no blue intrusion. My question: does ChimeraX incorporate any geometry-dependent information — bond angles, torsional state, idatm type reassignment based on local environment — into atom rendering or classification that would cause identical coloring commands to produce visually different results across structures? Or is ChimeraX rendering purely atomic positions as recorded in the mmCIF file with no geometry-based modification? Two Images attached Thank you for your help Wes Dennis ------------------------------
{kind=link}
{kind=link}

Hi Wes, Any representation of the atoms directly (stick, ball-and-stick, sphere) uses exactly the coordinates of the atoms as given in the input file. Only "abstracted" representations such as ribbons with smoothed paths might be away from the actual atomic coordinates given in the file. As to different coloring, it must be that the two files have different atom names or something like that -- the coloring will be applied exactly to whatever set of atoms you specified in your command. Coloring does not automatically relate to torsional stress or any other property. Sometimes (depending on molecule size) the default coloring when you initially display the molecule is by heteroatom (nitrogens blue, oxygens red, etc.) but if you actually gave a coloring command like "color yellow" it should do exactly as you say. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On May 14, 2026, at 6:59 PM, Wes Dennis via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi, My nameis Wes Dennis and I am a retired Geophysicist who is just interested in stuff. I am trying to understand what I am seeing with two ChimeraX images. I applied identical atom-name based coloring commands to the same residue range in two different crystal structures of the same molecule — backbone atoms colored blue, aromatic base atoms colored yellow. The visual result is strikingly different between the two structures. In one, blue color appears to spread into the base panels — a structure in which torsional stresses are present. In the other, where torsional stresses are absent, the base panels are clean yellow with no blue intrusion. My question: does ChimeraX incorporate any geometry-dependent information — bond angles, torsional state, idatm type reassignment based on local environment — into atom rendering or classification that would cause identical coloring commands to produce visually different results across structures? Or is ChimeraX rendering purely atomic positions as recorded in the mmCIF file with no geometry-based modification? Two Images attached
Thank you for your help
Wes Dennis <image - Copy.png><image (2) - Copy.png>

Hi All, I have a session with 80 homotetramers (4 x 1400 residues). I want to split all the tetramers, align all chains and calculate RMSF. I specially took precaution to disassociate all chains. Nevertheless, when I give the command
Dear All, I have a session with 80 homotetramers (4 x 1400 residues). I want to split all the tetramers, align all chains and calculate RMSF for all 320 chains. I specially took precaution to disassociate all chains. Nevertheless, when I give the command Select All split sel chains ChimeraX starts to associate all chains with all remaining chains, which takes >8 hours. Can you please suggest how to proceed? Best regards Sasha
Thank you for your rapid response. Subject: Follow-up: Color Bleeding Observation and MCP/Claude Integration Question Dear Elaine, Thank you again for your prompt and helpful response to my earlier question about atom display and coordinate rendering in ChimeraX. I wanted to follow up with two items. First, a follow-up on the color bleeding question. After your confirmation that ChimeraX renders atom positions using exact mmCIF coordinates, I ran a quantitative analysis to investigate what drove the visual difference between two sets of structures. Using glycosidic torsion angles (chi angles, defined as O4'-C1'-N9-C4 for purines and O4'-C1'-N1-C2 for pyrimidines) as an internal coordinate requiring no structural alignment, we found systematic differences of 20-30 degrees at specific residues between the two structural states. The visual color bleeding at the glycosidic junctions in the tier-colored images appears to reflect these real coordinate differences — the backbone atoms are physically displaced into the base panel visual space in one state but not the other. I wanted to share this in case it provides useful context for understanding of how the tier-coloring is a display with real physical meaning in ChimeraX, and to confirm that your assessment was correct and led directly to a productive result. Second, a question about Claude integration. I recently became aware that ChimeraX includes a built-in MCP command that may allow direct integration with Claude Desktop via Anthropic's Model Context Protocol. I am currently running ChimeraX 1.11.1 on Windows 11. Could you confirm whether MCP integration is supported in version 1.11.1, and if so, point me toward some setup documentation? I am now copying and pasting commands manually between Claude and ChimeraX, and a direct connection would .help a lot Thank you for your time and for the excellent software support your team provides. Best regards, Wes Dennis On Fri, May 15, 2026 at 10:25 AM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Wes, Any representation of the atoms directly (stick, ball-and-stick, sphere) uses exactly the coordinates of the atoms as given in the input file. Only "abstracted" representations such as ribbons with smoothed paths might be away from the actual atomic coordinates given in the file.
As to different coloring, it must be that the two files have different atom names or something like that -- the coloring will be applied exactly to whatever set of atoms you specified in your command. Coloring does not automatically relate to torsional stress or any other property. Sometimes (depending on molecule size) the default coloring when you initially display the molecule is by heteroatom (nitrogens blue, oxygens red, etc.) but if you actually gave a coloring command like "color yellow" it should do exactly as you say.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On May 14, 2026, at 6:59 PM, Wes Dennis via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Hi, My nameis Wes Dennis and I am a retired Geophysicist who is just interested in stuff. I am trying to understand what I am seeing with two ChimeraX images. I applied identical atom-name based coloring commands to the same residue range in two different crystal structures of the same molecule — backbone atoms colored blue, aromatic base atoms colored yellow. The visual result is strikingly different between the two structures. In one, blue color appears to spread into the base panels — a structure in which torsional stresses are present. In the other, where torsional stresses are absent, the base panels are clean yellow with no blue intrusion. My question: does ChimeraX incorporate any geometry-dependent information — bond angles, torsional state, idatm type reassignment based on local environment — into atom rendering or classification that would cause identical coloring commands to produce visually different results across structures? Or is ChimeraX rendering purely atomic positions as recorded in the mmCIF file with no geometry-based modification? Two Images attached
Thank you for your help
Wes Dennis <image - Copy.png><image (2) - Copy.png>
Dear Wes, Regarding Claude integration, I am not the expert on this topic, so perhaps the other developers will have additional comments. However, you can see an example of using Claude with ChimeraX here: <https://www.rbvi.ucsf.edu/chimerax/data/claude-dec2025/claude_mcp.html> ... as well as the "mcp" command help: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/mcp.html> Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On May 17, 2026, at 9:55 AM, Wes Dennis via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thank you for your rapid response.
Subject: Follow-up: Color Bleeding Observation and MCP/Claude Integration Question
Dear Elaine,
Thank you again for your prompt and helpful response to my earlier question about atom display and coordinate rendering in ChimeraX.
I wanted to follow up with two items.
First, a follow-up on the color bleeding question. After your confirmation that ChimeraX renders atom positions using exact mmCIF coordinates, I ran a quantitative analysis to investigate what drove the visual difference between two sets of structures. Using glycosidic torsion angles (chi angles, defined as O4'-C1'-N9-C4 for purines and O4'-C1'-N1-C2 for pyrimidines) as an internal coordinate requiring no structural alignment, we found systematic differences of 20-30 degrees at specific residues between the two structural states. The visual color bleeding at the glycosidic junctions in the tier-colored images appears to reflect these real coordinate differences — the backbone atoms are physically displaced into the base panel visual space in one state but not the other. I wanted to share this in case it provides useful context for understanding of how the tier-coloring is a display with real physical meaning in ChimeraX, and to confirm that your assessment was correct an! d led directly to a productive result.
Second, a question about Claude integration. I recently became aware that ChimeraX includes a built-in MCP command that may allow direct integration with Claude Desktop via Anthropic's Model Context Protocol. I am currently running ChimeraX 1.11.1 on Windows 11. Could you confirm whether MCP integration is supported in version 1.11.1, and if so, point me toward some setup documentation? I am now copying and pasting commands manually between Claude and ChimeraX, and a direct connection would .help a lot
Thank you for your time and for the excellent software support your team provides.
Best regards, Wes Dennis
On Fri, May 15, 2026 at 10:25 AM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Wes, Any representation of the atoms directly (stick, ball-and-stick, sphere) uses exactly the coordinates of the atoms as given in the input file. Only "abstracted" representations such as ribbons with smoothed paths might be away from the actual atomic coordinates given in the file.
As to different coloring, it must be that the two files have different atom names or something like that -- the coloring will be applied exactly to whatever set of atoms you specified in your command. Coloring does not automatically relate to torsional stress or any other property. Sometimes (depending on molecule size) the default coloring when you initially display the molecule is by heteroatom (nitrogens blue, oxygens red, etc.) but if you actually gave a coloring command like "color yellow" it should do exactly as you say.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On May 14, 2026, at 6:59 PM, Wes Dennis via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi, My nameis Wes Dennis and I am a retired Geophysicist who is just interested in stuff. I am trying to understand what I am seeing with two ChimeraX images. I applied identical atom-name based coloring commands to the same residue range in two different crystal structures of the same molecule — backbone atoms colored blue, aromatic base atoms colored yellow. The visual result is strikingly different between the two structures. In one, blue color appears to spread into the base panels — a structure in which torsional stresses are present. In the other, where torsional stresses are absent, the base panels are clean yellow with no blue intrusion. My question: does ChimeraX incorporate any geometry-dependent information — bond angles, torsional state, idatm type reassignment based on local environment — into atom rendering or classification that would cause identical coloring commands to produce visually different results across structures? Or is ChimeraX rendering purely atomic positions as recorded in the mmCIF file with no geometry-based modification? Two Images attached
Thank you for your help
Wes Dennis <image - Copy.png><image (2) - Copy.png>

Hi Sasha, I guess I don’t understand what you’re describing. Do you have sequence/alignment windows open? Are they necessary? Is there some reason you can’t just close them before you do all the splitting? There is a hack you can use to prevent auto-association, but you have to do it before opening the relevant alignments/sequences. Go to “Sequences” section of the ChimeraX Preferences/Settings and change the value for "Auto-associate if fewer than this many sequences” to 1. That will prevent any auto-association from happening for any sequences/alignments that you open while the value is 1. --Eric Eric Pettersen UCSF Computer Graphics Lab
On May 17, 2026, at 1:57 AM, Alexandra Zahradnikova via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear All,
I have a session with 80 homotetramers (4 x 1400 residues). I want to split all the tetramers, align all chains and calculate RMSF for all 320 chains. I specially took precaution to disassociate all chains. Nevertheless, when I give the command
Select All split sel chains
ChimeraX starts to associate all chains with all remaining chains, which takes >8 hours. Can you please suggest how to proceed?
Best regards Sasha _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Dear Eric, This is a file which was, previpisly aligned. The alignment was closed and it does not show in the Tools. But somehow it still persists. I do not know how to delete it. When I opened the file, there was an alignment present and the chains were associated. I dissociated them and closed the alignment. When I gave the Split command, the models were split but I was mot able to see the result because the alignment started running. I solved the problem by reading the cif files instead of opening the previous session file, but I would prefer to be able to delete the alignment. I will try the command that you suggested but I don't know if it will persist after I open the session file. Best regards Sasha Sent from my Galaxy -------- Original message --------From: Eric Pettersen <pett@cgl.ucsf.edu> Date: 18/05/2026 23:08 (GMT+01:00) To: Alexandra Zahradnikova <Alexandra.Zahradnikova@savba.sk> Cc: chimerax-users <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Association taking long time Hi Sasha, I guess I don’t understand what you’re describing. Do you have sequence/alignment windows open? Are they necessary? Is there some reason you can’t just close them before you do all the splitting? There is a hack you can use to prevent auto-association, but you have to do it before opening the relevant alignments/sequences. Go to “Sequences” section of the ChimeraX Preferences/Settings and change the value for "Auto-associate if fewer than this many sequences” to 1. That will prevent any auto-association from happening for any sequences/alignments that you open while the value is 1.--Eric Eric Pettersen UCSF Computer Graphics Lab> On May 17, 2026, at 1:57 AM, Alexandra Zahradnikova via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:> > Dear All,> > I have a session with 80 homotetramers (4 x 1400 residues). I want to split all the tetramers, align all chains and calculate RMSF for all 320 chains.> I specially took precaution to disassociate all chains. Nevertheless, when I give the command> > Select All> split sel chains> > ChimeraX starts to associate all chains with all remaining chains, which takes >8 hours.> Can you please suggest how to proceed? > > Best regards> Sasha> _______________________________________________> ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Dear Eric, I tried to follow your suggestion, i.e., opened the file and then I selected for "Auto-associate if fewer than this many sequences” to 2 (1 did not work). However, after splitting the models, they started to auto-associate despite this setting. Bets regards Sasha From: Alexandra Zahradnikova [mailto:alexandra.zahradnikova@savba.sk] Sent: Tuesday, May 19, 2026 07:05 To: chimerax-users@cgl.ucsf.edu Subject: Re: [chimerax-users] Association taking long time Dear Eric, This is a file which was, previpisly aligned. The alignment was closed and it does not show in the Tools. But somehow it still persists. I do not know how to delete it. When I opened the file, there was an alignment present and the chains were associated. I dissociated them and closed the alignment. When I gave the Split command, the models were split but I was mot able to see the result because the alignment started running. I solved the problem by reading the cif files instead of opening the previous session file, but I would prefer to be able to delete the alignment. I will try the command that you suggested but I don't know if it will persist after I open the session file. Best regards Sasha Sent from my Galaxy -------- Original message -------- From: Eric Pettersen <pett@cgl.ucsf.edu> Date: 18/05/2026 23:08 (GMT+01:00) To: Alexandra Zahradnikova <Alexandra.Zahradnikova@savba.sk> Cc: chimerax-users <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Association taking long time Hi Sasha, I guess I don’t understand what you’re describing. Do you have sequence/alignment windows open? Are they necessary? Is there some reason you can’t just close them before you do all the splitting? There is a hack you can use to prevent auto-association, but you have to do it before opening the relevant alignments/sequences. Go to “Sequences” section of the ChimeraX Preferences/Settings and change the value for "Auto-associate if fewer than this many sequences” to 1. That will prevent any auto-association from happening for any sequences/alignments that you open while the value is 1. --Eric Eric Pettersen UCSF Computer Graphics Lab
On May 17, 2026, at 1:57 AM, Alexandra Zahradnikova via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear All,
I have a session with 80 homotetramers (4 x 1400 residues). I want to split all the tetramers, align all chains and calculate RMSF for all 320 chains. I specially took precaution to disassociate all chains. Nevertheless, when I give the command
Select All split sel chains
ChimeraX starts to associate all chains with all remaining chains, which takes >8 hours. Can you please suggest how to proceed?
Best regards Sasha _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hi Sasha, After you close all the sequence/alignment windows, open ChimeraX's Python shell (Tools→General→Shell) and type: len(session.alignments) The output should be 0. If not, there are alignments open. If so, you may have to send me a session file from just before you split the structures, so I can give you better advice about what to do. --Eric
On May 19, 2026, at 8:18 AM, Alexandra Zahradnikova via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Eric, I tried to follow your suggestion, i.e., opened the file and then I selected for "Auto-associate if fewer than this many sequences” to 2 (1 did not work). However, after splitting the models, they started to auto-associate despite this setting. Bets regards Sasha
From: Alexandra Zahradnikova [mailto:alexandra.zahradnikova@savba.sk] Sent: Tuesday, May 19, 2026 07:05 To: chimerax-users@cgl.ucsf.edu Subject: Re: [chimerax-users] Association taking long time
Dear Eric, This is a file which was, previpisly aligned. The alignment was closed and it does not show in the Tools. But somehow it still persists. I do not know how to delete it.
When I opened the file, there was an alignment present and the chains were associated. I dissociated them and closed the alignment. When I gave the Split command, the models were split but I was mot able to see the result because the alignment started running.
I solved the problem by reading the cif files instead of opening the previous session file, but I would prefer to be able to delete the alignment. I will try the command that you suggested but I don't know if it will persist after I open the session file.
Best regards Sasha
Sent from my Galaxy
-------- Original message -------- From: Eric Pettersen <pett@cgl.ucsf.edu> Date: 18/05/2026 23:08 (GMT+01:00) To: Alexandra Zahradnikova <Alexandra.Zahradnikova@savba.sk> Cc: chimerax-users <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Association taking long time
Hi Sasha, I guess I don’t understand what you’re describing. Do you have sequence/alignment windows open? Are they necessary? Is there some reason you can’t just close them before you do all the splitting? There is a hack you can use to prevent auto-association, but you have to do it before opening the relevant alignments/sequences. Go to “Sequences” section of the ChimeraX Preferences/Settings and change the value for "Auto-associate if fewer than this many sequences” to 1. That will prevent any auto-association from happening for any sequences/alignments that you open while the value is 1.
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On May 17, 2026, at 1:57 AM, Alexandra Zahradnikova via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear All,
I have a session with 80 homotetramers (4 x 1400 residues). I want to split all the tetramers, align all chains and calculate RMSF for all 320 chains. I specially took precaution to disassociate all chains. Nevertheless, when I give the command
Select All split sel chains
ChimeraX starts to associate all chains with all remaining chains, which takes >8 hours. Can you please suggest how to proceed?
Best regards Sasha _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
participants (5)
-
 Alexandra Zahradnikova
Alexandra Zahradnikova -
Alexandra Zahradnikova
-
 Elaine Meng
Elaine Meng -
 Eric Pettersen
Eric Pettersen -
 Wes Dennis
Wes Dennis