
Hello, I would appreciate if you please answer the following questions: 1. I am using chimeraX, and I have a trouble getting results from Alphafold. The Google Colab keep on termination without saving results. Could you please help? 2. I am using hydrogen bond tool, how can I save all the H-bonds in the protein as file containing all the information for the paired residues involved in the H bond. Similarly, I also would like to save the information of residues involved in the protein-protein interactions. I hope to hear from you. Best, Amita Department of Biology University of Pennsylvania

Hi Amita, You would need to give a more detailed explanation of what you mean by "Google Colab keep on termination". Does Google Colab disconnect? Does an error message from AlphaFold appear? Does it finish and say "Downloading results..."? Tom
On Apr 2, 2024, at 12:58 PM, Amita Mohan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I would appreciate if you please answer the following questions:
1. I am using chimeraX, and I have a trouble getting results from Alphafold. The Google Colab keep on termination without saving results. Could you please help?
2. I am using hydrogen bond tool, how can I save all the H-bonds in the protein as file containing all the information for the paired residues involved in the H bond. Similarly, I also would like to save the information of residues involved in the protein-protein interactions.
I hope to hear from you.
Best,
Amita Department of Biology University of Pennsylvania _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/

Dear Amita, (1) The AlphaFold calculation is on Google Colab which has limitations, and it may or may not be possible to run the calculation that you are trying to run. It depends on your specific data, and what the reason was for termination. See the help for the possible problems and possible solutions: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html#caveats> (2) There is an option to save a file in the H-Bonds tool (and also the hbonds command). H-Bonds tool has "Write information to" option, you can turn this on and choose file, see the bottom of the help page: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/hbonds.html> "hbonds" command has "saveFile" option, see the help: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/hbonds.html#saveFile> I don't know which tool you are using to find protein-protein interactions. If you are using Contacts, just like H-Bonds, it has a "Write information to" option, and the "contacts" command has a "saveFile" option. <https://rbvi.ucsf.edu/chimerax/docs/user/tools/clashes.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/clashes.html#saveFile> Or, if you are using ChimeraX menu: Select... Contacts, after you make the selection <https://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html> ... then you can write a list of the selected atoms to a file using command "info atoms sel" or "info residues sel" if you also use the "saveFile" option in that command, for example on my Mac I can use command: info residues sel saveFile ~/Desktop/residue-list See the help <https://rbvi.ucsf.edu/chimerax/docs/user/commands/info.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/info.html#saveFile> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 2, 2024, at 12:58 PM, Amita Mohan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I would appreciate if you please answer the following questions:
1. I am using chimeraX, and I have a trouble getting results from Alphafold. The Google Colab keep on termination without saving results. Could you please help?
2. I am using hydrogen bond tool, how can I save all the H-bonds in the protein as file containing all the information for the paired residues involved in the H bond. Similarly, I also would like to save the information of residues involved in the protein-protein interactions.
I hope to hear from you.
Best, Amita Department of Biology University of Pennsylvania
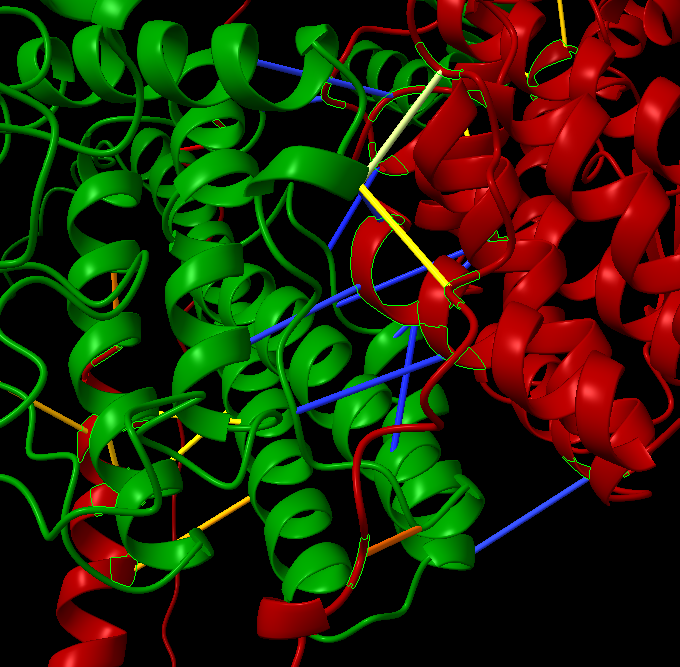
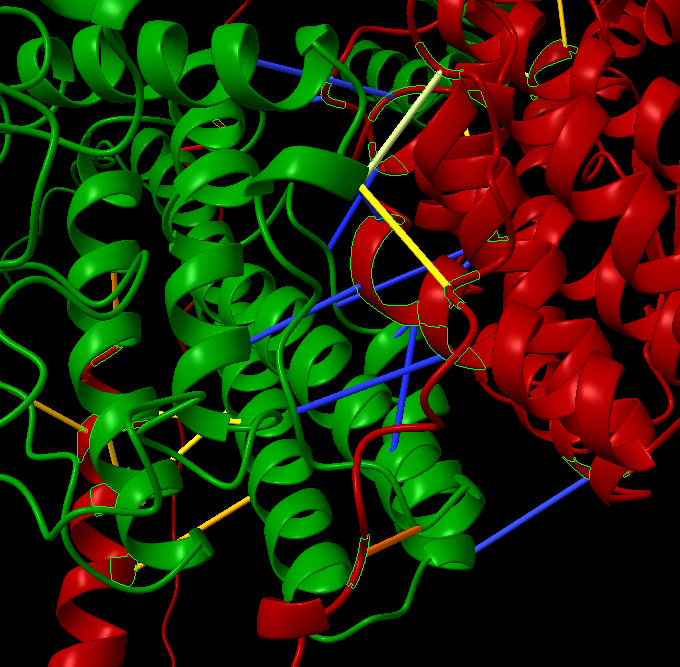
Hello Elaine, I have a question, in chain A and chain B, I have identified the contact resides using the command ‘alphafold contacts /A to /B distance 3’. I want to write the chain A and B (for example /A Asn 431 — /B The 321) contact residues as file. How can also get the information on the blue and yellow interactions as well. I used the information you have provided in previous email, I can write only either chain but not the contact residue information that will help in protein-protein interactions. The example is provided in the below screen shot. Thank you. Amita
On Apr 2, 2024, at 4:30 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
info residues sel saveFile ~/Desktop/residue-list 
{kind=link}
Hi Amita, First be aware that "alphafold contacts" is not primarily meant to identify contacts but instead to show with colored lines the alphafold confidence values (PAE) in the relative positions of pairs of residues. The colors of the lines show the PAE values. The "distance 3" part merely limits where the lines are drawn to residue pairs that are close together. If you still want that information, given the description above, you can use the "outputFile" option of "alphafold contacts" to write a list of the residue pairs and their closest distance, as described in the help: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/alphafold.html#contacts> This outputFile option was also described in a previous post: <https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/...> However, in general: If you just want to identify the atomic contacts in any structure regardless of whether it is from alphafold or not, it may be better to use the menu: Select... Contacts... , see: <https://rbvi.ucsf.edu/chimerax/docs/user/selectcontacts.html> This approach uses either the "contacts" command or "interfaces" command depending on the options you choose, and you could also use either of those commands directly instead of via the menu. The "contacts" command has a "saveFile" option to write a list of the contacting atom pairs and their distances. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/clashes.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/interfaces.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
Begin forwarded message:
From: Amita Mohan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Subject: [chimerax-users] Re: ChimeraX question Date: April 16, 2024 at 9:18:18 AM PDT To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Reply-To: Amita Mohan <amimohan@sas.upenn.edu>
Hello Elaine,
I have a question, in chain A and chain B, I have identified the contact resides using the command ‘alphafold contacts /A to /B distance 3’. I want to write the chain A and B (for example /A Asn 431 — /B The 321) contact residues as file. How can also get the information on the blue and yellow interactions as well. I used the information you have provided in previous email, I can write only either chain but not the contact residue information that will help in protein-protein interactions. The example is provided in the below screen shot.
Thank you.
Amita
On Apr 2, 2024, at 4:30 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
info residues sel saveFile ~/Desktop/residue-list 
{kind=link}
participants (3)
-
 Amita Mohan
Amita Mohan -
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard