Chai discovery data

Hello, I have downloaded the Chai discovery data in .npy and .json file formats. How can I open these files in Chimera X? Thank you Reza

Hi Reza, if you mean Chai the machine learning model/alphafold clone then you'd have to do a bit of work in connecting their output with Chimerax. .npy files are generally the format in which python's `numpy` package saves multidimensional arrays to disk. I'm not sure what their json output is. Your best bet is probably finding how the data in your files relates to chimerax' api and writing a script to connect the two. On Sat, Jan 25, 2025 at 4:36 PM Reza Salavati, Prof. via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I have downloaded the Chai discovery data in .npy and .json file formats.
How can I open these files in Chimera X?
Thank you
Reza
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/

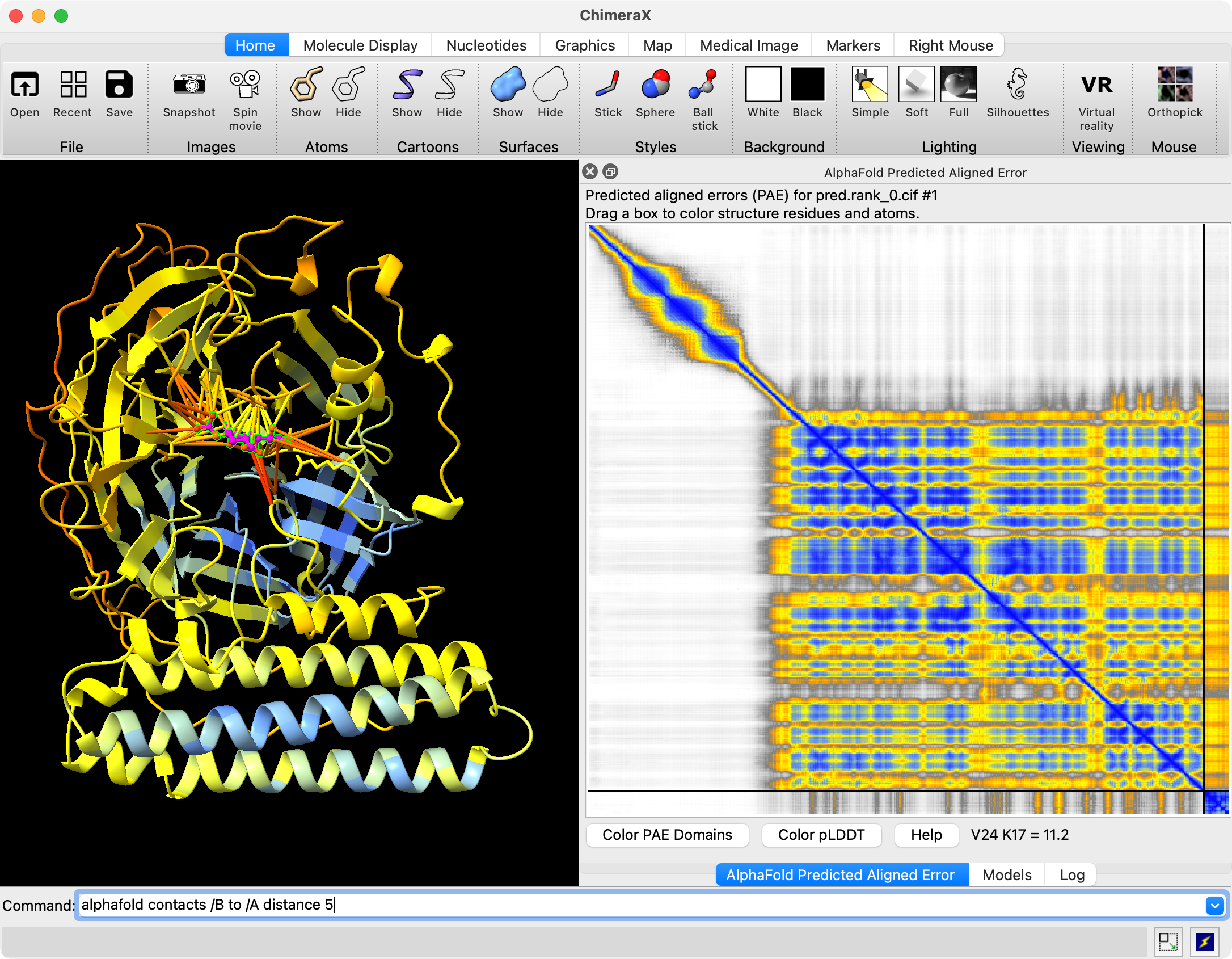
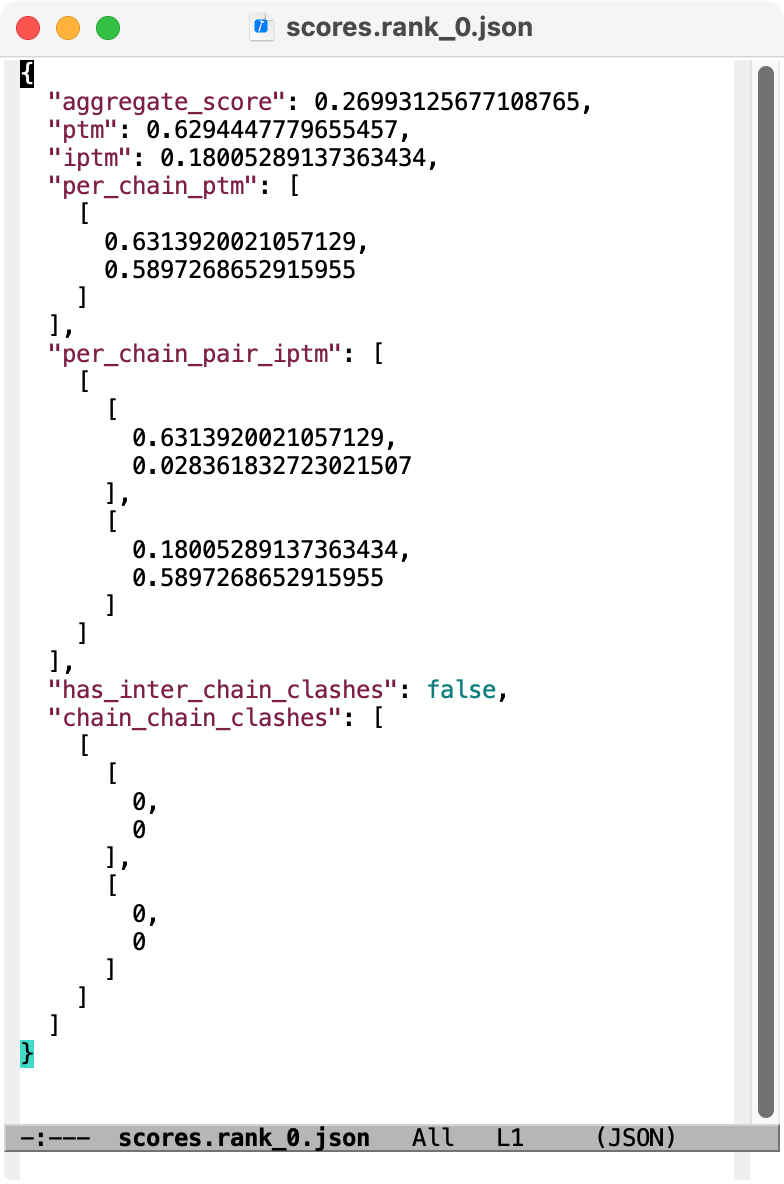
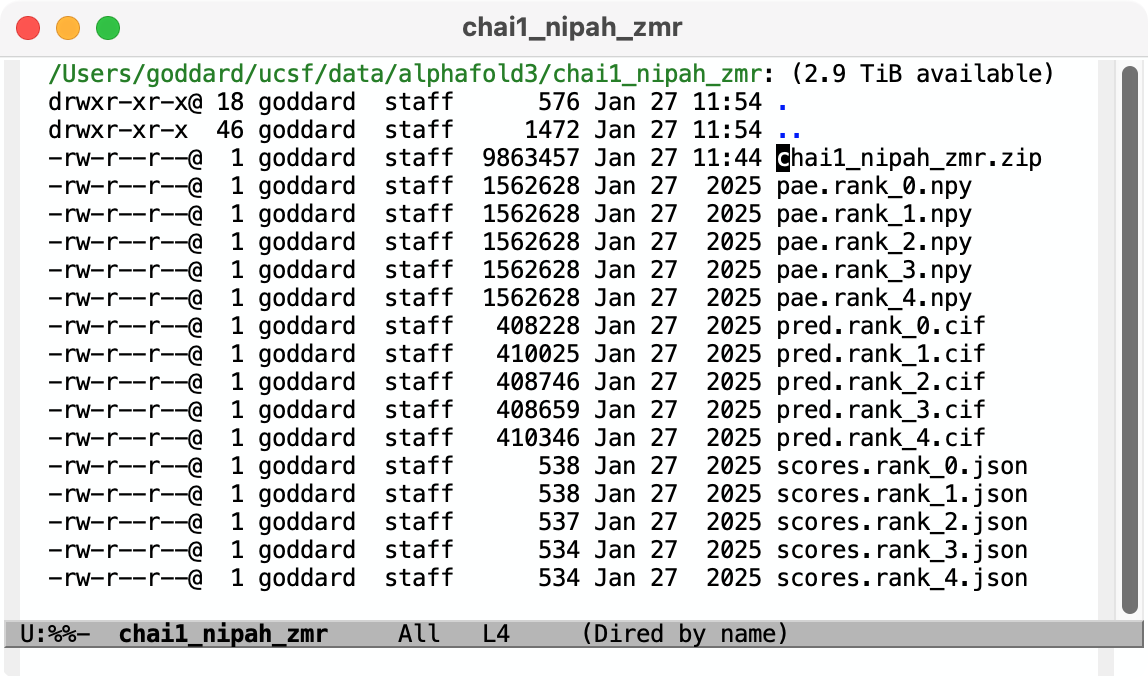
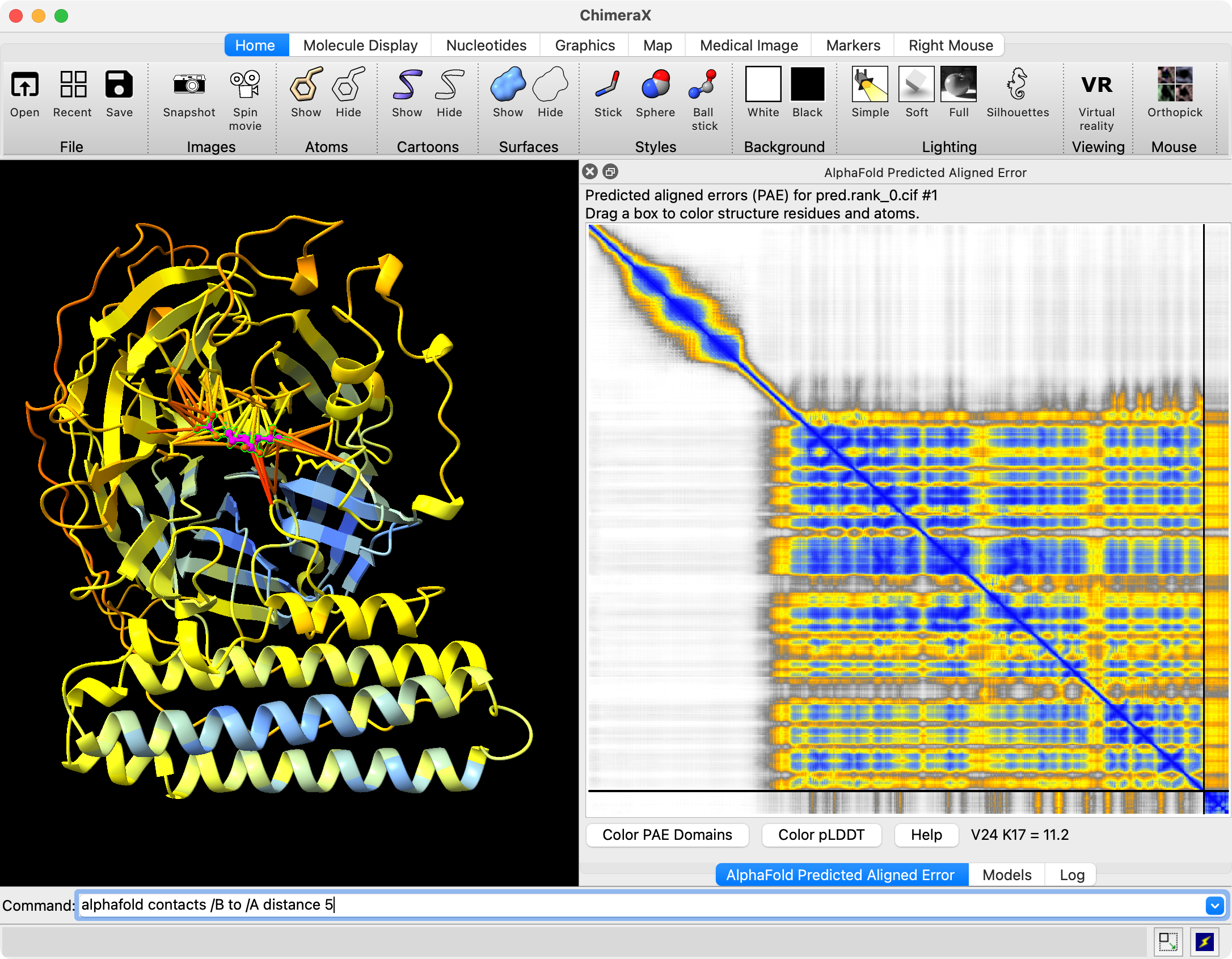
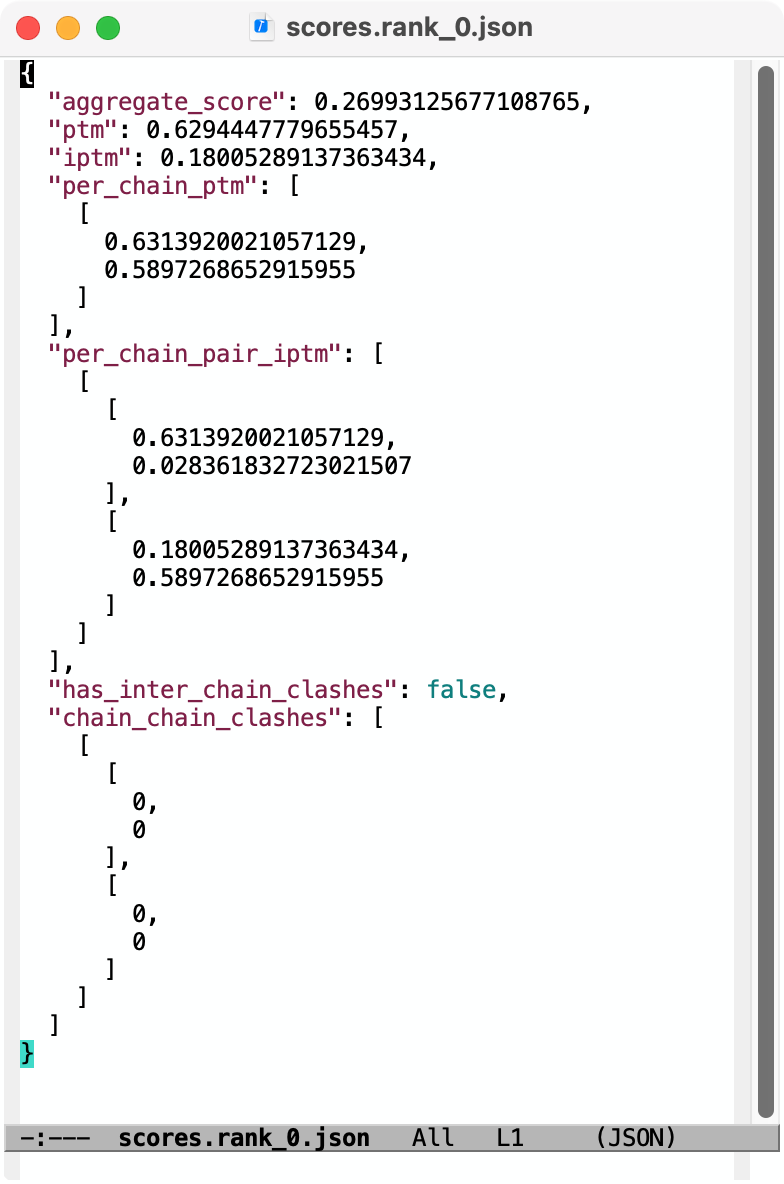
Hi Reza, Chai-1 outputs predicted aligned error (PAE) in .npy format. You can open it in ChimeraX 1.9 by first opening the predicted structure pred.rank_0.cif using the File / Open dialog. Then use menu Tools / Structure Prediction / AlphaFold Error Plot to open the PAE file pae.rank_0.npy. This shows the PAE as a heat map as shown in the image below. Use current ChimeraX version 1.9 not 1.8 since Chai-1 didn't exist when 1.8 was released and so the numpy PAE format was only added in 1.9. For an explanation how to visualize that on the structure see this description https://www.rbvi.ucsf.edu/chimerax/data/pae-apr2022/pae.html The scores.rank_0.json file contains summary confidence scores (per-chain pTM, between chain ipTM, ...) and clashes on a whole model or per-chain level. ChimeraX doesn't have any tool to visualize those but you can show them in any text editor, image below. Tom Chai-1 predicted structure and PAE plot shown in ChimeraX 1.9 (Nipah virus G protein with Zanamivir (pink) bound, low confidence).  Chai-1 scores.rank_0.json shown in a text editor  Files returned by Chai-1 server 
On Jan 25, 2025, at 3:11 PM, Artie Kushner via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Reza, if you mean Chai the machine learning model/alphafold clone then you'd have to do a bit of work in connecting their output with Chimerax. .npy files are generally the format in which python's `numpy` package saves multidimensional arrays to disk. I'm not sure what their json output is. Your best bet is probably finding how the data in your files relates to chimerax' api and writing a script to connect the two.
On Sat, Jan 25, 2025 at 4:36 PM Reza Salavati, Prof. via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hello,
I have downloaded the Chai discovery data in .npy and .json file formats.
How can I open these files in Chimera X?
Thank you
Reza
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
{kind=link}
{kind=link}
Hi Tom Thank you for getting back to me. It was very helpful. Can the files you open be easily manipulated by selecting amino acids on the sequence or structure? Is there a way to superimpose and compare a protein bound with two different ligands? Best Reza From: Tom Goddard <goddard@sonic.net> Sent: January 27, 2025 3:39 PM To: Reza Salavati, Prof. <reza.salavati@mcgill.ca> Cc: chimerax-users@cgl.ucsf.edu; Artie Kushner <rtkushner@gmail.com> Subject: Re: [chimerax-users] Chai discovery data Hi Reza, Chai-1 outputs predicted aligned error (PAE) in .npy format. You can open it in ChimeraX 1.9 by first opening the predicted structure pred.rank_0.cif using the File / Open dialog. Then use menu Tools / Structure Prediction / AlphaFold Error Plot to open the PAE file pae.rank_0.npy. This shows the PAE as a heat map as shown in the image below. Use current ChimeraX version 1.9 not 1.8 since Chai-1 didn't exist when 1.8 was released and so the numpy PAE format was only added in 1.9. For an explanation how to visualize that on the structure see this description https://www.rbvi.ucsf.edu/chimerax/data/pae-apr2022/pae.html The scores.rank_0.json file contains summary confidence scores (per-chain pTM, between chain ipTM, ...) and clashes on a whole model or per-chain level. ChimeraX doesn't have any tool to visualize those but you can show them in any text editor, image below. Tom Chai-1 predicted structure and PAE plot shown in ChimeraX 1.9 (Nipah virus G protein with Zanamivir (pink) bound, low confidence). [cid:image001.png@01DB7269.E5145890] Chai-1 scores.rank_0.json shown in a text editor [cid:image002.png@01DB7269.E5145890] Files returned by Chai-1 server [cid:image003.png@01DB7269.E5145890] On Jan 25, 2025, at 3:11 PM, Artie Kushner via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hi Reza, if you mean Chai the machine learning model/alphafold clone then you'd have to do a bit of work in connecting their output with Chimerax. .npy files are generally the format in which python's `numpy` package saves multidimensional arrays to disk. I'm not sure what their json output is. Your best bet is probably finding how the data in your files relates to chimerax' api and writing a script to connect the two. On Sat, Jan 25, 2025 at 4:36 PM Reza Salavati, Prof. via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hello, I have downloaded the Chai discovery data in .npy and .json file formats. How can I open these files in Chimera X? Thank you Reza _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu<mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu<mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
{kind=link}
{kind=link}

Hi Reza, In general, not necessarily related to Chai-1 or any prediction methods, you can show the sequence of a protein in the Sequence Viewer, and selections in one (sequence or 3D structure) are reflected in the other. Show chain sequence with menu: Tools... Sequence... Show Sequence Viewer, or you can open sequences from UniProt, or sequence alignment files, etc. that will associate with your structure if the sequence is similar enough. See Sequence Viewer help: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/sequenceviewer.html> Also in general, you can superimpose protein structures with the Matchmaker tool (in menu under Tools... Structure Analysis) or "matchmaker" command to have it figure out the residue-residue correspondences for you, or "align" command if you want to specify exactly which pairs of atoms to use. See help pages: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/matchmaker.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/align.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 29, 2025, at 1:21 PM, Reza Salavati, Prof. via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Tom Thank you for getting back to me. It was very helpful. Can the files you open be easily manipulated by selecting amino acids on the sequence or structure? Is there a way to superimpose and compare a protein bound with two different ligands? Best Reza
participants (4)
-
 Artie Kushner
Artie Kushner -
 Elaine Meng
Elaine Meng -
 Reza Salavati, Prof.
Reza Salavati, Prof. -
 Tom Goddard
Tom Goddard