Re: [Chimera-users] Chimera X menu "View" vs. "Focus"

Hi Christian, We named it “View” instead of “Focus” because it issues a “view” command, but your points have merit — “Focus” is more evocative of what the action actually does, and is less confusable with hide/show, so after some discussion we have renamed the menu item to “Focus” and the change will be in the next daily build. The menu item item still issues/logs a “view” command to do its work. We are aware that the direct equivalent of the “Fetch By ID” dialog is missing in ChimeraX and we intend to add it. It’s just a matter of implementation priorities, so it could still be a little while — but it’s useful to know that it’s definitely missed, which will make it more likely to get worked on sooner. Also, there is a list specifically for ChimeraX user issues — chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>, (cc’ed here), which would be better to discuss ChimeraX topics on than chimera-users… --Eric Eric Pettersen UCSF Computer Graphics Lab
On Sep 25, 2023, at 12:47 AM, C. Klein via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Dear Chimera Team
first of all, thank you very much for your important and good work on the Chimera software.
Two comments:
I did some recent work with Chimera X. The menu item "Focus" was changed to "View", which I find less suitable. The term "View" is often applied in other contexts, in particular if a certain residue should be shown vs. not shown. In contrast, "Focus" is not ambiguous, it is not related to showing vs. hiding certain parts of the molecular representation but exactly indicates what is done by the command.
The direct pdb download from rcsb or whatever repository is a highly useful tool which is more difficult to access than it used to be. Ok I know it is there, I type "open xxxx" and I use the command line quite a lot anyway, but for others who start with the menus it does not appear to be accessible. Perhaps I also missed something, but then others may not find it as well.
Thank you and best wishes Christian Klein _______________________________________________ Chimera-users mailing list -- chimera-users@cgl.ucsf.edu To unsubscribe send an email to chimera-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimera-users@cgl.ucsf.edu/

Cosigned re fetch by id being missed - particularly for EMDB (maps & map+model), which I don’t see an obvious way to access currently even on the command line (apologies if I missed it and it is obvious!) Cheers Oli
On Sep 25, 2023, at 1:11 PM, Eric Pettersen via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Christian, We named it “View” instead of “Focus” because it issues a “view” command, but your points have merit — “Focus” is more evocative of what the action actually does, and is less confusable with hide/show, so after some discussion we have renamed the menu item to “Focus” and the change will be in the next daily build. The menu item item still issues/logs a “view” command to do its work. We are aware that the direct equivalent of the “Fetch By ID” dialog is missing in ChimeraX and we intend to add it. It’s just a matter of implementation priorities, so it could still be a little while — but it’s useful to know that it’s definitely missed, which will make it more likely to get worked on sooner. Also, there is a list specifically for ChimeraX user issues — chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>, (cc’ed here), which would be better to discuss ChimeraX topics on than chimera-users…
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Sep 25, 2023, at 12:47 AM, C. Klein via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Dear Chimera Team
first of all, thank you very much for your important and good work on the Chimera software.
Two comments:
I did some recent work with Chimera X. The menu item "Focus" was changed to "View", which I find less suitable. The term "View" is often applied in other contexts, in particular if a certain residue should be shown vs. not shown. In contrast, "Focus" is not ambiguous, it is not related to showing vs. hiding certain parts of the molecular representation but exactly indicates what is done by the command.
The direct pdb download from rcsb or whatever repository is a highly useful tool which is more difficult to access than it used to be. Ok I know it is there, I type "open xxxx" and I use the command line quite a lot anyway, but for others who start with the menus it does not appear to be accessible. Perhaps I also missed something, but then others may not find it as well.
Thank you and best wishes Christian Klein _______________________________________________ Chimera-users mailing list -- chimera-users@cgl.ucsf.edu To unsubscribe send an email to chimera-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimera-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/

Hi Oli, The open command can be used for all kinds of fetches, e.g. open emdb:1024 - or - open 1024 from emdb open pubchem:4056 open smiles:C1CCCN1 open alphafold:LDLR_HUMAN etc. See fetch from online resources: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#fetch> But as far as I know there isn't a map+model fetch. If I remember correctly there were some issues (alignment?) and I don't know if EMDB entries have a standardized approach for that situation. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 25, 2023, at 10:16 AM, Oliver Clarke via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Cosigned re fetch by id being missed - particularly for EMDB (maps & map+model), which I don’t see an obvious way to access currently even on the command line (apologies if I missed it and it is obvious!)
Cheers Oli
On Sep 25, 2023, at 1:11 PM, Eric Pettersen via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Christian, We named it “View” instead of “Focus” because it issues a “view” command, but your points have merit — “Focus” is more evocative of what the action actually does, and is less confusable with hide/show, so after some discussion we have renamed the menu item to “Focus” and the change will be in the next daily build. The menu item item still issues/logs a “view” command to do its work. We are aware that the direct equivalent of the “Fetch By ID” dialog is missing in ChimeraX and we intend to add it. It’s just a matter of implementation priorities, so it could still be a little while — but it’s useful to know that it’s definitely missed, which will make it more likely to get worked on sooner. Also, there is a list specifically for ChimeraX user issues — chimerax-users@cgl.ucsf.edu, (cc’ed here), which would be better to discuss ChimeraX topics on than chimera-users…
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Sep 25, 2023, at 12:47 AM, C. Klein via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Dear Chimera Team
first of all, thank you very much for your important and good work on the Chimera software.
Two comments:
I did some recent work with Chimera X. The menu item "Focus" was changed to "View", which I find less suitable. The term "View" is often applied in other contexts, in particular if a certain residue should be shown vs. not shown. In contrast, "Focus" is not ambiguous, it is not related to showing vs. hiding certain parts of the molecular representation but exactly indicates what is done by the command.
The direct pdb download from rcsb or whatever repository is a highly useful tool which is more difficult to access than it used to be. Ok I know it is there, I type "open xxxx" and I use the command line quite a lot anyway, but for others who start with the menus it does not appear to be accessible. Perhaps I also missed something, but then others may not find it as well.
Thank you and best wishes Christian Klein

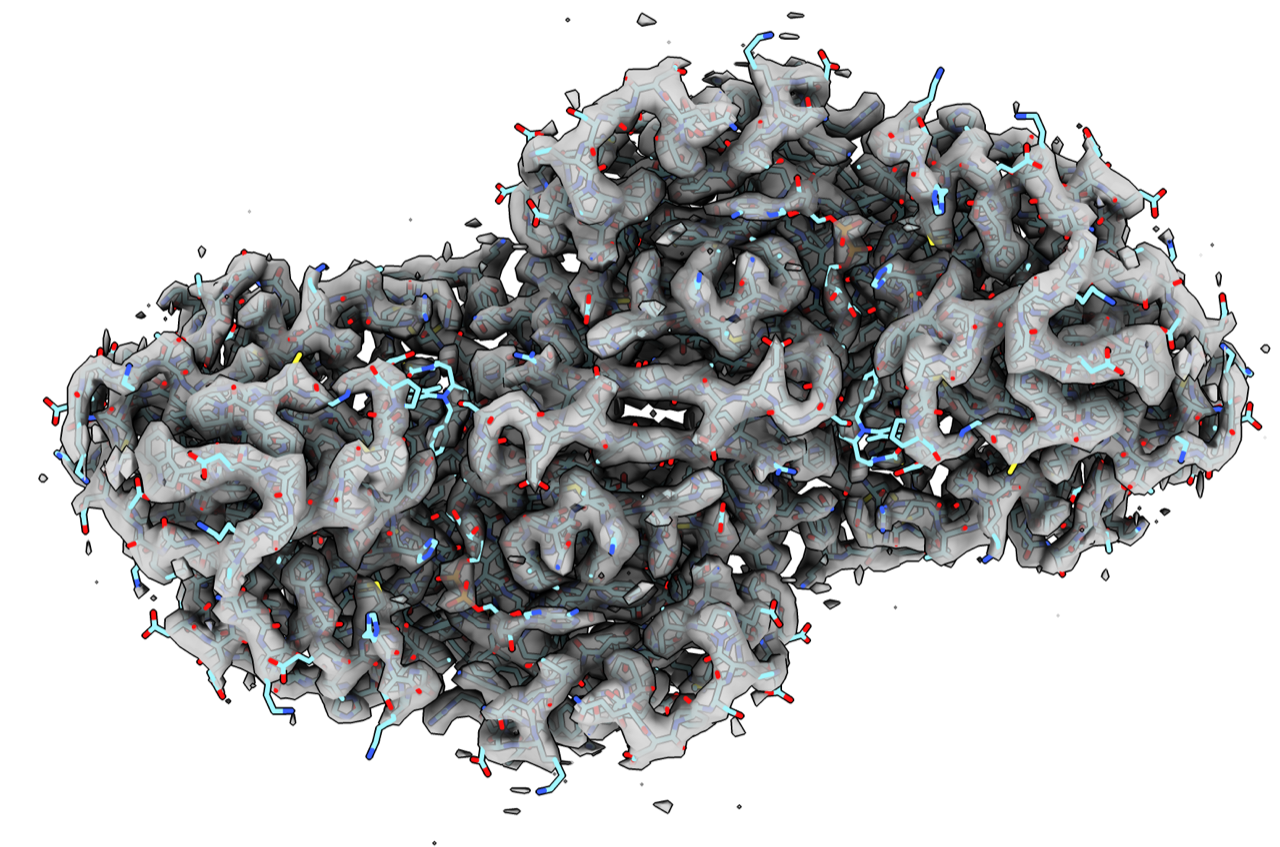
Hi Oli, I added options to fetch the fit PDB atomic models when fetching a map using a new "fits true" option to the open command, or the new emdb_fits database name. These 3 equivalent commands fetch both the EMDB map and the fit atomic model: open 34282 from emdb fits true open 34282 from emdb_fits open emdb_fits:34282 The EMDB fetching code now fetches both the map and the EMDB metadata file (e.g. emdb-34282.xml) and they are cached in your Downloads directory ~/Downloads/ChimeraX/EMDB Even if you don't ask for the fit models it still fetches the meta-data and reports the PDB ids of the fit models in the Log as links and clicking on those links opens the atomic models. open 34828 from emdb > Opened emdb 34282 as #1, grid size 180,180,180, pixel 1.1, shown at level 0.7, step 1, values float32, fit PDB 8gv3 <cxcmd: open 8gv3> Since ChimeraX now fetches the EMDB meta-data I made it also use the author suggested contour level as the initial surface level. Hopefully that will usually be a better surface level than the heuristic one ChimeraX used to use. By the way, the old Chimera Fetch by ID method for getting both the EMDB maps and fit atomic models stopped working some time ago when EMDB changed the way the fit PDBs were listed in the XML meta-data file. We won't be fixing Chimera since it only gets critical maintenance, so best to use ChimeraX. Tom 
On Sep 25, 2023, at 12:00 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Oli, The open command can be used for all kinds of fetches, e.g.
open emdb:1024 - or - open 1024 from emdb
open pubchem:4056
open smiles:C1CCCN1
open alphafold:LDLR_HUMAN
etc.
See fetch from online resources: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#fetch>
But as far as I know there isn't a map+model fetch. If I remember correctly there were some issues (alignment?) and I don't know if EMDB entries have a standardized approach for that situation.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 25, 2023, at 10:16 AM, Oliver Clarke via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Cosigned re fetch by id being missed - particularly for EMDB (maps & map+model), which I don’t see an obvious way to access currently even on the command line (apologies if I missed it and it is obvious!)
Cheers Oli
On Sep 25, 2023, at 1:11 PM, Eric Pettersen via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Christian, We named it “View” instead of “Focus” because it issues a “view” command, but your points have merit — “Focus” is more evocative of what the action actually does, and is less confusable with hide/show, so after some discussion we have renamed the menu item to “Focus” and the change will be in the next daily build. The menu item item still issues/logs a “view” command to do its work. We are aware that the direct equivalent of the “Fetch By ID” dialog is missing in ChimeraX and we intend to add it. It’s just a matter of implementation priorities, so it could still be a little while — but it’s useful to know that it’s definitely missed, which will make it more likely to get worked on sooner. Also, there is a list specifically for ChimeraX user issues — chimerax-users@cgl.ucsf.edu, (cc’ed here), which would be better to discuss ChimeraX topics on than chimera-users…
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Sep 25, 2023, at 12:47 AM, C. Klein via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Dear Chimera Team
first of all, thank you very much for your important and good work on the Chimera software.
Two comments:
I did some recent work with Chimera X. The menu item "Focus" was changed to "View", which I find less suitable. The term "View" is often applied in other contexts, in particular if a certain residue should be shown vs. not shown. In contrast, "Focus" is not ambiguous, it is not related to showing vs. hiding certain parts of the molecular representation but exactly indicates what is done by the command.
The direct pdb download from rcsb or whatever repository is a highly useful tool which is more difficult to access than it used to be. Ok I know it is there, I type "open xxxx" and I use the command line quite a lot anyway, but for others who start with the menus it does not appear to be accessible. Perhaps I also missed something, but then others may not find it as well.
Thank you and best wishes Christian Klein
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
Wonderful thank you Tom! Setting the contour from the metadata is a nice touch too, that is very convenient! Cheers Oli
On Sep 26, 2023, at 9:22 PM, Tom Goddard <goddard@sonic.net> wrote:
Hi Oli,
I added options to fetch the fit PDB atomic models when fetching a map using a new "fits true" option to the open command, or the new emdb_fits database name. These 3 equivalent commands fetch both the EMDB map and the fit atomic model:
open 34282 from emdb fits true open 34282 from emdb_fits open emdb_fits:34282
The EMDB fetching code now fetches both the map and the EMDB metadata file (e.g. emdb-34282.xml) and they are cached in your Downloads directory
~/Downloads/ChimeraX/EMDB
Even if you don't ask for the fit models it still fetches the meta-data and reports the PDB ids of the fit models in the Log as links and clicking on those links opens the atomic models.
open 34828 from emdb > Opened emdb 34282 as #1, grid size 180,180,180, pixel 1.1, shown at level 0.7, step 1, values float32, fit PDB 8gv3 <cxcmd: open 8gv3>
Since ChimeraX now fetches the EMDB meta-data I made it also use the author suggested contour level as the initial surface level. Hopefully that will usually be a better surface level than the heuristic one ChimeraX used to use.
By the way, the old Chimera Fetch by ID method for getting both the EMDB maps and fit atomic models stopped working some time ago when EMDB changed the way the fit PDBs were listed in the XML meta-data file. We won't be fixing Chimera since it only gets critical maintenance, so best to use ChimeraX.
Tom <emdb_34272_pdb_8gv3.png>
On Sep 25, 2023, at 12:00 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Oli, The open command can be used for all kinds of fetches, e.g.
open emdb:1024 - or - open 1024 from emdb
open pubchem:4056
open smiles:C1CCCN1
open alphafold:LDLR_HUMAN
etc.
See fetch from online resources: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#fetch>
But as far as I know there isn't a map+model fetch. If I remember correctly there were some issues (alignment?) and I don't know if EMDB entries have a standardized approach for that situation.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 25, 2023, at 10:16 AM, Oliver Clarke via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Cosigned re fetch by id being missed - particularly for EMDB (maps & map+model), which I don’t see an obvious way to access currently even on the command line (apologies if I missed it and it is obvious!)
Cheers Oli
On Sep 25, 2023, at 1:11 PM, Eric Pettersen via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Christian, We named it “View” instead of “Focus” because it issues a “view” command, but your points have merit — “Focus” is more evocative of what the action actually does, and is less confusable with hide/show, so after some discussion we have renamed the menu item to “Focus” and the change will be in the next daily build. The menu item item still issues/logs a “view” command to do its work. We are aware that the direct equivalent of the “Fetch By ID” dialog is missing in ChimeraX and we intend to add it. It’s just a matter of implementation priorities, so it could still be a little while — but it’s useful to know that it’s definitely missed, which will make it more likely to get worked on sooner. Also, there is a list specifically for ChimeraX user issues — chimerax-users@cgl.ucsf.edu, (cc’ed here), which would be better to discuss ChimeraX topics on than chimera-users…
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Sep 25, 2023, at 12:47 AM, C. Klein via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Dear Chimera Team
first of all, thank you very much for your important and good work on the Chimera software.
Two comments:
I did some recent work with Chimera X. The menu item "Focus" was changed to "View", which I find less suitable. The term "View" is often applied in other contexts, in particular if a certain residue should be shown vs. not shown. In contrast, "Focus" is not ambiguous, it is not related to showing vs. hiding certain parts of the molecular representation but exactly indicates what is done by the command.
The direct pdb download from rcsb or whatever repository is a highly useful tool which is more difficult to access than it used to be. Ok I know it is there, I type "open xxxx" and I use the command line quite a lot anyway, but for others who start with the menus it does not appear to be accessible. Perhaps I also missed something, but then others may not find it as well.
Thank you and best wishes Christian Klein
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/

Dear Oliver Dear All great, just downloaded the latest version! Extremely useful "fetch" utility! Works like a charm. Thank you and keep up the good work Christian On 9/25/2023 7:16 PM, Oliver Clarke wrote:
Cosigned re fetch by id being missed - particularly for EMDB (maps & map+model), which I don’t see an obvious way to access currently even on the command line (apologies if I missed it and it is obvious!)
Cheers Oli
On Sep 25, 2023, at 1:11 PM, Eric Pettersen via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Christian, We named it “View” instead of “Focus” because it issues a “view” command, but your points have merit — “Focus” is more evocative of what the action actually does, and is less confusable with hide/show, so after some discussion we have renamed the menu item to “Focus” and the change will be in the next daily build. The menu item item still issues/logs a “view” command to do its work. We are aware that the direct equivalent of the “Fetch By ID” dialog is missing in ChimeraX and we intend to add it. It’s just a matter of implementation priorities, so it could still be a little while — but it’s useful to know that it’s definitely missed, which will make it more likely to get worked on sooner. Also, there is a list specifically for ChimeraX user issues — chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>, (cc’ed here), which would be better to discuss ChimeraX topics on than chimera-users…
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Sep 25, 2023, at 12:47 AM, C. Klein via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Dear Chimera Team
first of all, thank you very much for your important and good work on the Chimera software.
Two comments:
I did some recent work with Chimera X. The menu item "Focus" was changed to "View", which I find less suitable. The term "View" is often applied in other contexts, in particular if a certain residue should be shown vs. not shown. In contrast, "Focus" is not ambiguous, it is not related to showing vs. hiding certain parts of the molecular representation but exactly indicates what is done by the command.
The direct pdb download from rcsb or whatever repository is a highly useful tool which is more difficult to access than it used to be. Ok I know it is there, I type "open xxxx" and I use the command line quite a lot anyway, but for others who start with the menus it does not appear to be accessible. Perhaps I also missed something, but then others may not find it as well.
Thank you and best wishes Christian Klein _______________________________________________ Chimera-users mailing list -- chimera-users@cgl.ucsf.edu To unsubscribe send an email to chimera-users-leave@cgl.ucsf.edu Archives: https://urldefense.proofpoint.com/v2/url?u=https-3A__mail.cgl.ucsf.edu_mailm...
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://urldefense.proofpoint.com/v2/url?u=https-3A__mail.cgl.ucsf.edu_mailm...
-- Prof. Dr. C. Klein; c.klein(at)https://urldefense.proofpoint.com/v2/url?u=http-3A__uni-2Dheidelberg.de&d=Dw... Medicinal Chemistry Institute of Pharmacy and Molecular Biotechnology IPMB Heidelberg University, INF 364 D-69120 Heidelberg Germany Phone: ++49-6221-54-4875 FAX : ++49-6221-54-6430
participants (5)
-
 C. Klein
C. Klein -
 Elaine Meng
Elaine Meng -
 Eric Pettersen
Eric Pettersen -
 Oliver Clarke
Oliver Clarke -
 Tom Goddard
Tom Goddard