ISOLDE and secondary structure restraints

Hi all, I just started model building in cryoEM maps and get great results using ISOLDE on AlphaFold2-derived initial models. I have a question which is mainly directed to Tristan I guess: Is is possible to use the command line/script to apply secondary structure restraints in ISOLDE? I would like to restrain the initial secondary structure assignment from Alphafold especially at resolution worse than 3.5 A. At the moment I select each and every residue range assigned to helix, parallel or antiparallel beta-strand one after the other and turn on the corresponding restrains in the ISOLDE GUI. Is there a better way via command line of script? Cheers, Guido -- PD Dr. Guido Hansen Group Leader UNIVERSITÄT ZU LÜBECK INSTITUT FÜR BIOCHEMIE Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de www.biochem.uni-luebeck.de [1] Ratzeburger Allee 160 23562 Lübeck Links: ------ [1] http://www.biochem.uni-luebeck.de

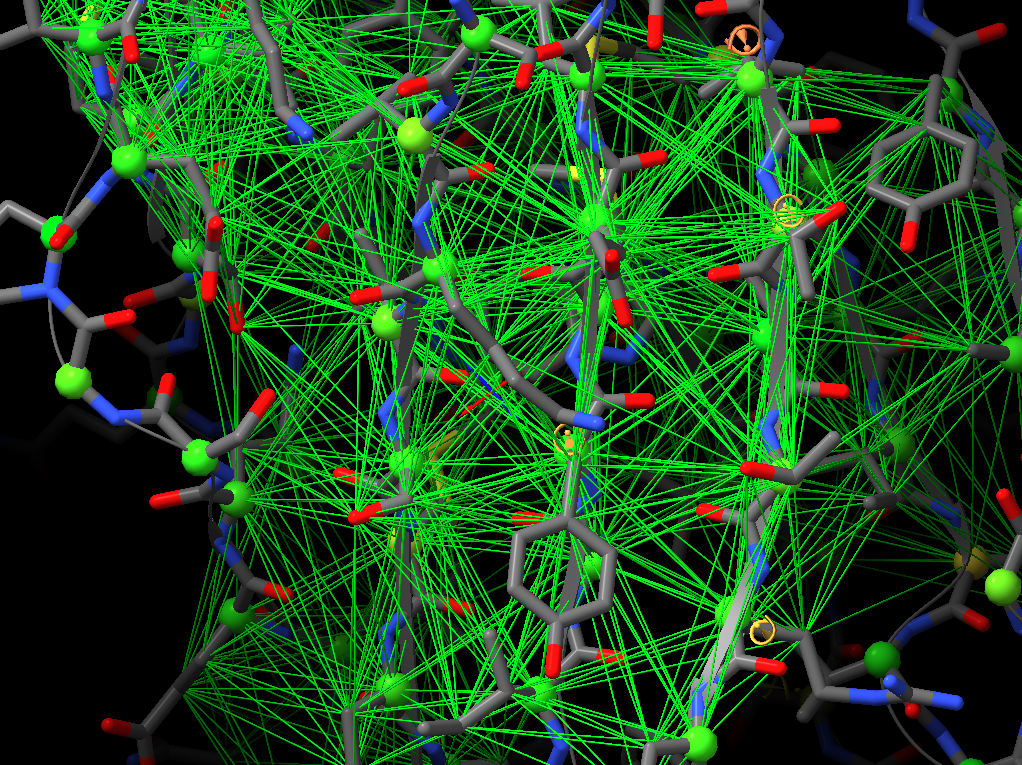
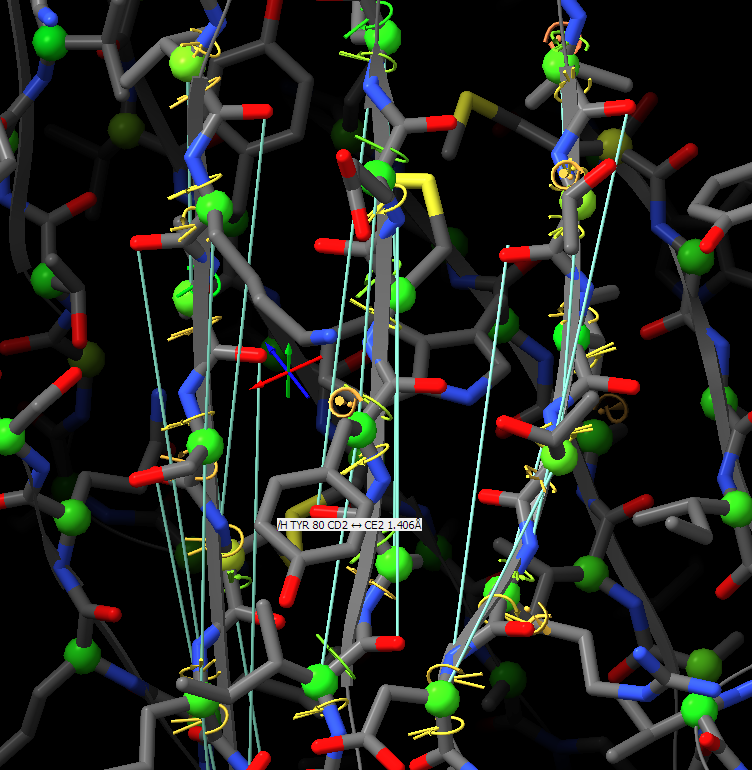
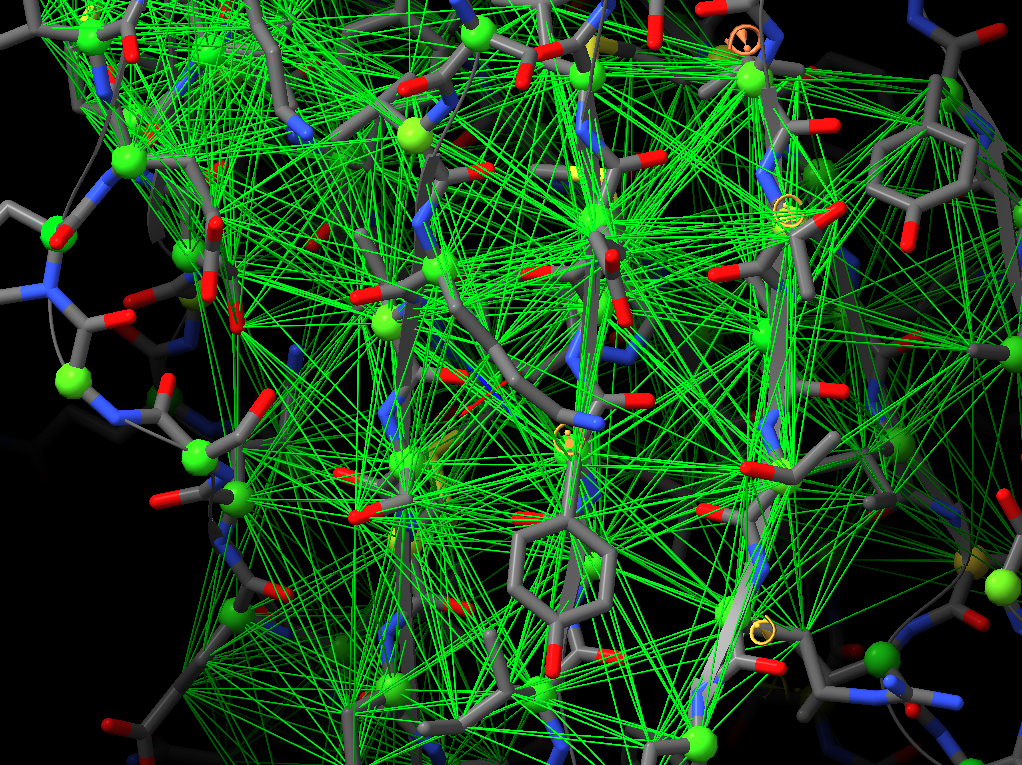
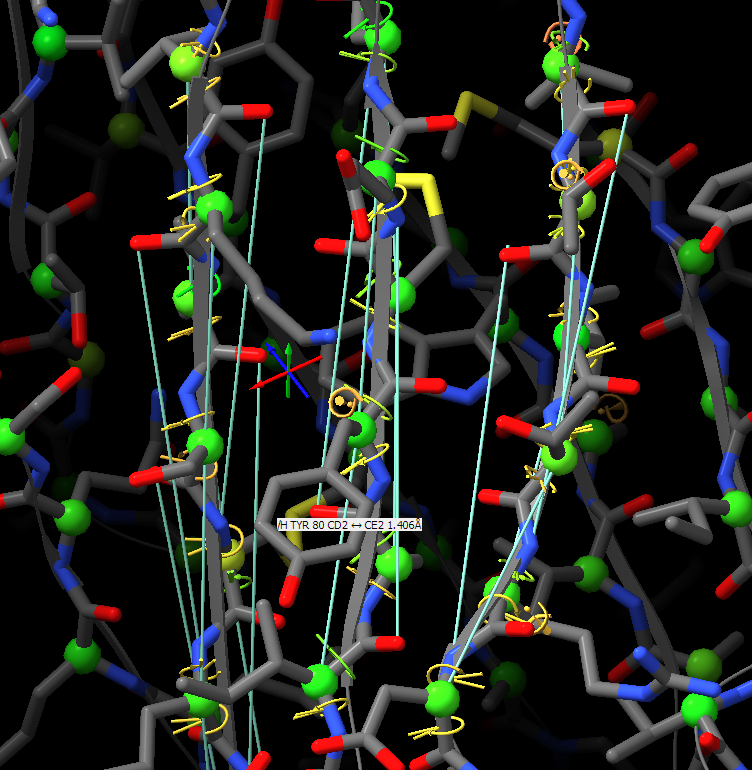
Hi, I would like to pick up this old thread. Do I interpret this correctly that ISOLDE ignores the secondary structure annotations in the PDB header and has its own way of calculating what’s a helix and a strand? If so, are these definitions written in the PDB header when the file is saved, or is there an option to do so? Many thanks, Matthias From: ChimeraX-users <chimerax-users-bounces@cgl.ucsf.edu> on behalf of Tristan Croll via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Date: Tuesday, 15. March 2022 at 17:30 To: Guido Hansen <hansen@biochem.uni-luebeck.de> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] ISOLDE and secondary structure restraints Oops! My bad - was responding on the move, and forgetting my own implementation. You're right, the "isolde restrain distances" command works on a per-residue basis, so sidechain atoms will be included regardless. On the other hand, the "isolde release distances" command works per-atom, so after the restrain command you can selectively release the sidechain restraints with: isolde release distances #1&sideonly ________________________________ From: Guido Hansen <hansen@biochem.uni-luebeck.de> Sent: 15 March 2022 16:20 To: Tristan Croll <tic20@cam.ac.uk> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] ISOLDE and secondary structure restraints Thanks for your suggestions. However, I think that when applying distance restraints using the command below also distances of mainchain atoms of secondary structure elements to nearby sidechain atoms get restrainted. isolde restrain distances #1&(helix|strand)&backbone [cid:part1.09p0uiHA.41fj6uxZ@biochem.uni-luebeck.de] What you get when using the gui is something like that: [cid:part2.0mNP8JTc.JBo6Z60y@biochem.uni-luebeck.de] -- Guido Am 13.03.2022 um 16:07 schrieb Tristan Croll: Not the hard-coded helix and beta-sheet restraints, I’m afraid. But you can get a very similar (probably better, TBH) result with something like: isolde restrain distances #1&(helix|strand)&backbone isolde restrain torsions #1&(helix|strand) sidechain false The advantage of those is that (a) they’ll work a bit better when the true conformation isn’t *perfect* helix or strand, and (b) they have a lot more flexibility in terms of your ability to fine-tune their behaviour (for models fetched from the Alphafold DB, try adding the argument “adjustForConfidence true” to automatically take the pLDDT and PAE values into account). — Tristan On 13 Mar 2022, at 14:42, Guido Hansen via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hi all, I just started model building in cryoEM maps and get great results using ISOLDE on AlphaFold2-derived initial models. I have a question which is mainly directed to Tristan I guess: Is is possible to use the command line/script to apply secondary structure restraints in ISOLDE? I would like to restrain the initial secondary structure assignment from Alphafold especially at resolution worse than 3.5 A. At the moment I select each and every residue range assigned to helix, parallel or antiparallel beta-strand one after the other and turn on the corresponding restrains in the ISOLDE GUI. Is there a better way via command line of script? Cheers, Guido -- PD Dr. Guido Hansen Group Leader [Image removed by sender. Uni-Lübeck] Universität zu Lübeck Institut für Biochemie Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de<mailto:hansen@biochem.uni-luebeck.de> www.biochem.uni-luebeck.de<http://www.biochem.uni-luebeck.de> Ratzeburger Allee 160 23562 Lübeck _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu<mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users -- PD Dr. Guido Hansen Group Leader [Image removed by sender. Uni-Lübeck] Universität zu Lübeck Institut für Biochemie Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de<mailto:hansen@biochem.uni-luebeck.de> www.biochem.uni-luebeck.de<http://www.biochem.uni-luebeck.de> Ratzeburger Allee 160 23562 Lübeck
{kind=link}
{kind=link}

Hi Matthias, The “(helix|strand)” selectors are core ChimeraX rather than ISOLDE. Beyond that, the answer is “it depends”. As I understand it, if the model has HELIX or STRAND records (or the mmCIF equivalents) then ChimeraX will respect those when first loading the model. If they don’t exist it will use dssp to calculate them. If dssp is used subsequently it will recalculate the secondary structure based on the current conformation - as far as I know once that’s happened there’s no way to reinstate the original definitions. Best, Tristan On Fri, 17 Nov 2023 at 11:19, Vorländer,Matthias Kopano via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Hi,
I would like to pick up this old thread. Do I interpret this correctly that ISOLDE ignores the secondary structure annotations in the PDB header and has its own way of calculating what’s a helix and a strand? If so, are these definitions written in the PDB header when the file is saved, or is there an option to do so?
Many thanks,
Matthias
*From: *ChimeraX-users <chimerax-users-bounces@cgl.ucsf.edu> on behalf of Tristan Croll via ChimeraX-users <chimerax-users@cgl.ucsf.edu> *Date: *Tuesday, 15. March 2022 at 17:30 *To: *Guido Hansen <hansen@biochem.uni-luebeck.de> *Cc: *chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> *Subject: *Re: [chimerax-users] ISOLDE and secondary structure restraints
Oops! My bad - was responding on the move, and forgetting my own implementation. You're right, the "isolde restrain distances" command works on a per-residue basis, so sidechain atoms will be included regardless. On the other hand, the "isolde release distances" command works per-atom, so after the restrain command you can selectively release the sidechain restraints with:
isolde release distances #1&sideonly ------------------------------
*From:* Guido Hansen <hansen@biochem.uni-luebeck.de> *Sent:* 15 March 2022 16:20 *To:* Tristan Croll <tic20@cam.ac.uk> *Cc:* chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> *Subject:* Re: [chimerax-users] ISOLDE and secondary structure restraints
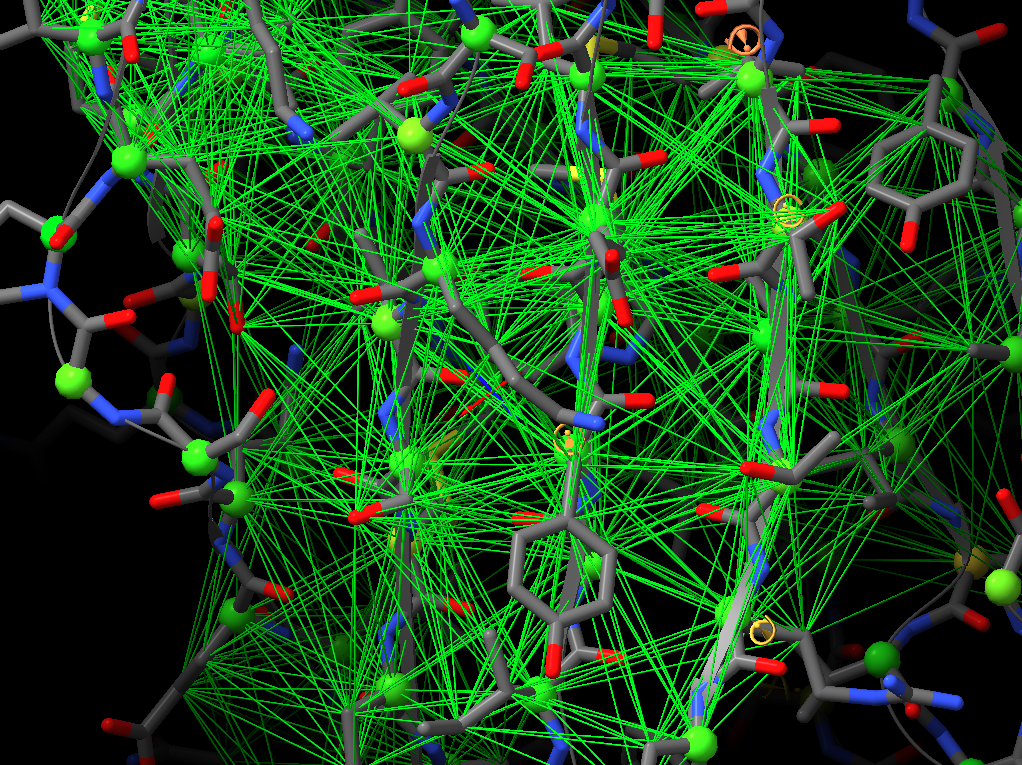
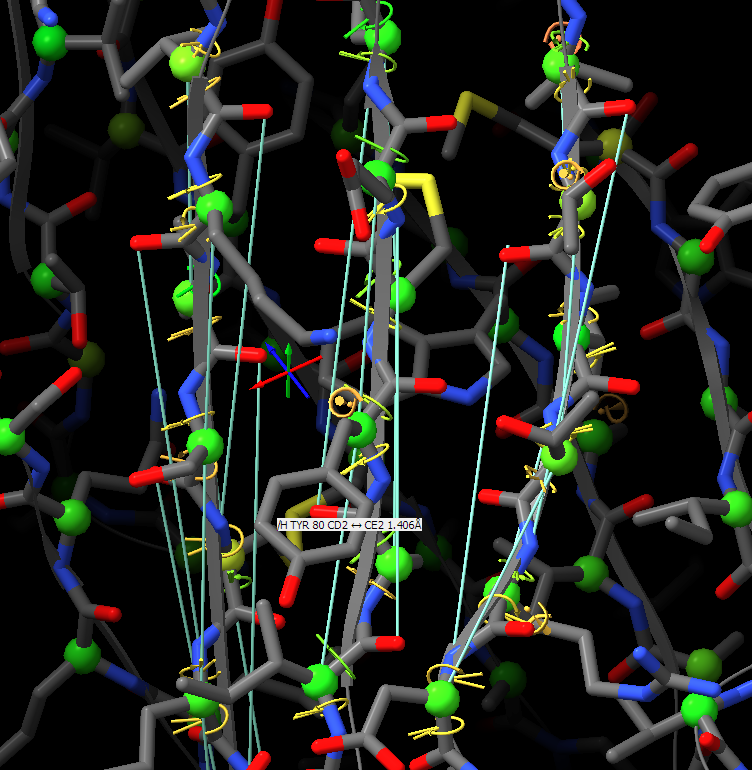
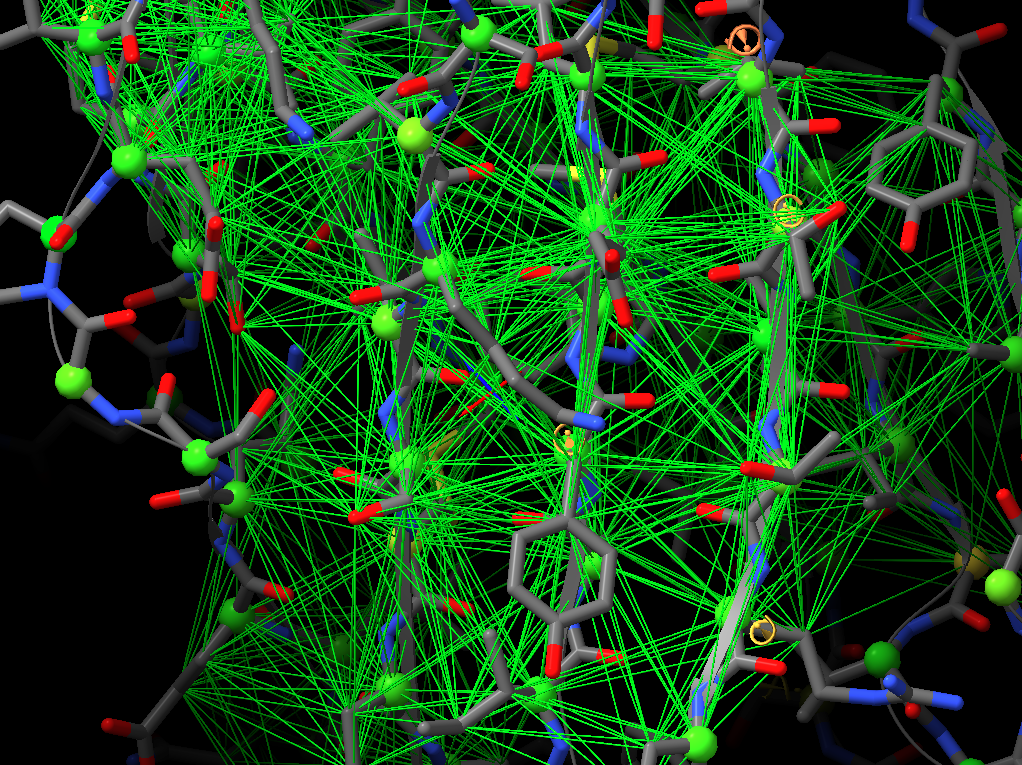
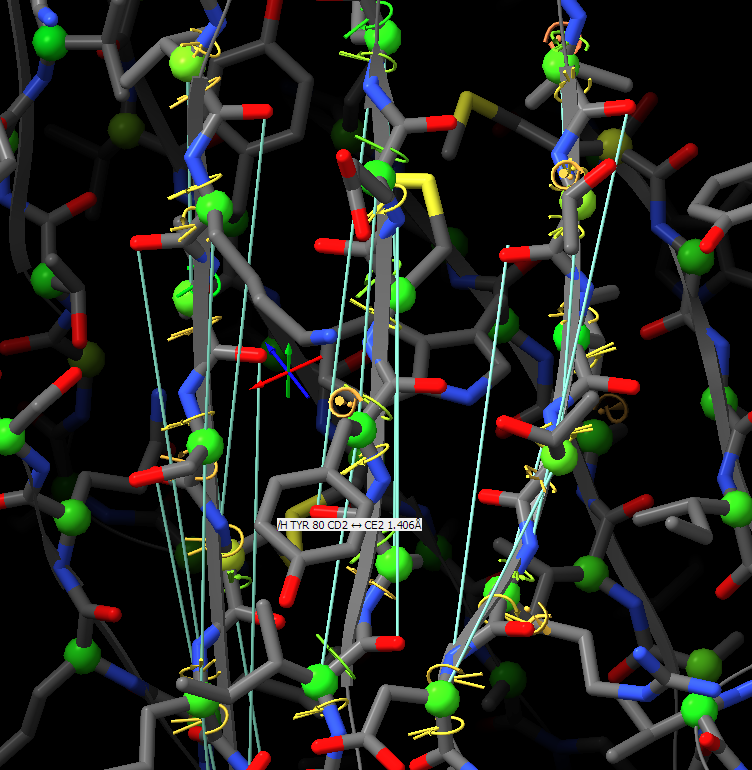
Thanks for your suggestions. However, I think that when applying distance restraints using the command below also distances of mainchain atoms of secondary structure elements to nearby sidechain atoms get restrainted.
isolde restrain distances #1&(helix|strand)&backbone
What you get when using the gui is something like that:
-- Guido
Am 13.03.2022 um 16:07 schrieb Tristan Croll:
Not the hard-coded helix and beta-sheet restraints, I’m afraid. But you can get a very similar (probably better, TBH) result with something like:
isolde restrain distances #1&(helix|strand)&backbone
isolde restrain torsions #1&(helix|strand) sidechain false
The advantage of those is that (a) they’ll work a bit better when the true conformation isn’t *perfect* helix or strand, and (b) they have a lot more flexibility in terms of your ability to fine-tune their behaviour (for models fetched from the Alphafold DB, try adding the argument “adjustForConfidence true” to automatically take the pLDDT and PAE values into account).
— Tristan
On 13 Mar 2022, at 14:42, Guido Hansen via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Hi all,
I just started model building in cryoEM maps and get great results using ISOLDE on AlphaFold2-derived initial models. I have a question which is mainly directed to Tristan I guess:
Is is possible to use the command line/script to apply secondary structure restraints in ISOLDE? I would like to restrain the initial secondary structure assignment from Alphafold especially at resolution worse than 3.5 A. At the moment I select each and every residue range assigned to helix, parallel or antiparallel beta-strand one after the other and turn on the corresponding restrains in the ISOLDE GUI. Is there a better way via command line of script?
Cheers, Guido
--
*PD Dr. Guido Hansen* Group Leader
[image: Image removed by sender. Uni-Lübeck]
*Universität zu Lübeck* *Institut für Biochemie*
Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de www.biochem.uni-luebeck.de
Ratzeburger Allee 160 <https://www.google.com/maps/search/Ratzeburger+Allee+160+%0D%0A23562+L%C3%BC...> 23562 Lübeck <https://www.google.com/maps/search/Ratzeburger+Allee+160+%0D%0A23562+L%C3%BC...>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
--
*PD Dr. Guido Hansen*
Group Leader
[image: Image removed by sender. Uni-Lübeck]
*Universität zu Lübeck* *Institut für Biochemie*
Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de www.biochem.uni-luebeck.de
Ratzeburger Allee 160 <https://www.google.com/maps/search/Ratzeburger+Allee+160+%0D%0A23562+L%C3%BC...> 23562 Lübeck <https://www.google.com/maps/search/Ratzeburger+Allee+160+%0D%0A23562+L%C3%BC...> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
-- Altos Labs UK Limited | England | Company reg 13484917 Registered address: 3rd Floor 1 Ashley Road, Altrincham, Cheshire, United Kingdom, WA14 2DT
{kind=link}
{kind=link}

Dear Tristan, I have a similar question then I exhume that topic... As I'm dealing with a ~1500 residues protein that I want to make dance in a density. Then as it is large I would like to use command lines to assign secondary structure to known helices and B-sheets instead of clicking on each residues to say this is an helix and this is a sheet. Is there a way to say to Isolde something like: 20 to 30 is an helix, 50 to 56 is a parallel sheet, ..., 1452 to 1460 is and helix. Please place your tensors and angles and lets dance?... Thank you for your answer. Michel

Hi Michel, I thought of a quick answer, though forgive me that I'm not Tristan. Have you tried first running the ChimeraX dssp command to assign the secondary structure, and next, choosing one of the secondary structure restraints buttons in the ISOLDE GUI's Restraints tab? -- Dan Richman, PhD (he/him/his) Research Associate, James Berger Lab Dept of Biophysics and Biophysical Chemistry Johns Hopkins University School of Medicine mobile: 201-669-0967 ________________________________ From: michel.thepaut--- via ChimeraX-users <chimerax-users@cgl.ucsf.edu> Sent: Wednesday, February 25, 2026 6:15 AM To: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: [chimerax-users] Re: ISOLDE and secondary structure restraints External Email - Use Caution Dear Tristan, I have a similar question then I exhume that topic... As I'm dealing with a ~1500 residues protein that I want to make dance in a density. Then as it is large I would like to use command lines to assign secondary structure to known helices and B-sheets instead of clicking on each residues to say this is an helix and this is a sheet. Is there a way to say to Isolde something like: 20 to 30 is an helix, 50 to 56 is a parallel sheet, ..., 1452 to 1460 is and helix. Please place your tensors and angles and lets dance?... Thank you for your answer. Michel _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://nam02.safelinks.protection.outlook.com/?url=https%3A%2F%2Fmail.cgl.ucsf.edu%2Fmailman%2Farchives%2Flist%2Fchimerax-users%40cgl.ucsf.edu%2F&data=05%7C02%7Cd.e.richman%40jhu.edu%7C86c14a4715a841a3908a08de7482066f%7C9fa4f438b1e6473b803f86f8aedf0dec%7C0%7C0%7C639076299049509513%7CUnknown%7CTWFpbGZsb3d8eyJFbXB0eU1hcGkiOnRydWUsIlYiOiIwLjAuMDAwMCIsIlAiOiJXaW4zMiIsIkFOIjoiTWFpbCIsIldUIjoyfQ%3D%3D%7C0%7C%7C%7C&sdata=ktoCJWIvzmm8TE5Q7X8oNRleEyaGvE7iM6z9fWpzDXQ%3D&reserved=0<https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/>
So... there currently isn't an option in ISOLDE to do this via the command line. But it's pretty easy to add. Unfortunately I *just* missed adding it into the ISOLDE 1.11 release, but you can add it to your session by putting the below code in a file called something like "isolde_restr_ss_cmd.py" and opening that in ChimeraX: ``` def restrain_ss(session, residues, ss_type): from chimerax.atomic import Residue structures = residues.unique_structures for s in structures: sresidues = s.residues.intersect(residues) strands = sresidues[sresidues.ss_types==Residue.SS_STRAND] helices = sresidues[sresidues.ss_types==Residue.SS_HELIX] from chimerax.isolde.restraints.restraint_utils import restrain_secondary_structure if ss_type == 'current': restrain_secondary_structure(session, strands, 'strand') restrain_secondary_structure(session, helices, 'helix') if ss_type == 'strand': restrain_secondary_structure(session, sresidues, 'strand') if ss_type == 'helix': restrain_secondary_structure(session, sresidues, 'helix') from chimerax.core.commands import CmdDesc, register, EnumOf from chimerax.atomic import ResiduesArg desc = CmdDesc( synopsis='Add backbone distance and torsion restraints to restrain residues to idealised secondary structure geometry', required=[ ('residues', ResiduesArg), ('ss_type', EnumOf(('strand', 'helix', 'current'))) ], ) register('isolde restrain ss', desc, restrain_ss, logger=session.logger) ``` This adds the command "isolde restrain ss {sel} {strand|helix|current}". The "strand" option will restrain everything in the selection to beta-strand (including restraining H-bonds to adjacent strands in a sheet if they're also part of the selection); "helix" will restrain it all to helix; "current" will restrain each residue to its currently-assigned type. Obviously you want to be very careful with the first two in particular (results will be fun but extremely "interesting" if you accidentally restrain the entire structure to helix!). Hope this helps, Tristan On Wed, Feb 25, 2026 at 3:56 PM Dan Richman via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Hi Michel, I thought of a quick answer, though forgive me that I'm not Tristan. Have you tried first running the ChimeraX dssp command to assign the secondary structure, and next, choosing one of the secondary structure restraints buttons in the ISOLDE GUI's Restraints tab?
-- Dan Richman, PhD (he/him/his) Research Associate, James Berger Lab Dept of Biophysics and Biophysical Chemistry Johns Hopkins University School of Medicine mobile: 201-669-0967 ------------------------------ *From:* michel.thepaut--- via ChimeraX-users <chimerax-users@cgl.ucsf.edu> *Sent:* Wednesday, February 25, 2026 6:15 AM *To:* chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> *Subject:* [chimerax-users] Re: ISOLDE and secondary structure restraints
External Email - Use Caution
Dear Tristan, I have a similar question then I exhume that topic...
As I'm dealing with a ~1500 residues protein that I want to make dance in a density. Then as it is large I would like to use command lines to assign secondary structure to known helices and B-sheets instead of clicking on each residues to say this is an helix and this is a sheet. Is there a way to say to Isolde something like: 20 to 30 is an helix, 50 to 56 is a parallel sheet, ..., 1452 to 1460 is and helix. Please place your tensors and angles and lets dance?...
Thank you for your answer. Michel _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://nam02.safelinks.protection.outlook.com/?url=https%3A%2F%2Fmail.cgl.u... <https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
-- Altos Labs UK Limited | England | Company reg 13484917 Registered address: 3rd Floor 1 Ashley Road, Altrincham, Cheshire, United Kingdom, WA14 2DT
(sending again because apparently putting Python code in-line triggers some security filter... apologies to those who get this twice). So... there currently isn't an option in ISOLDE to do this via the command line. But it's pretty easy to add. Unfortunately I *just* missed adding it into the ISOLDE 1.11 release, but you can add it to your session by renaming the attached file to "restrain_ss_cmd.py" and opening that in ChimeraX. This adds the command "isolde restrain ss {sel} {strand|helix|current}". The "strand" option will restrain everything in the selection to beta-strand (including restraining H-bonds to adjacent strands in a sheet if they're also part of the selection); "helix" will restrain it all to helix; "current" will restrain each residue to its currently-assigned type. Obviously you want to be very careful with the first two in particular (results will be fun but extremely "interesting" if you accidentally restrain the entire structure to helix or strand!). Hope this helps, Tristan On Wed, Feb 25, 2026 at 3:56 PM Dan Richman via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Hi Michel, I thought of a quick answer, though forgive me that I'm not Tristan. Have you tried first running the ChimeraX dssp command to assign the secondary structure, and next, choosing one of the secondary structure restraints buttons in the ISOLDE GUI's Restraints tab?
-- Dan Richman, PhD (he/him/his) Research Associate, James Berger Lab Dept of Biophysics and Biophysical Chemistry Johns Hopkins University School of Medicine mobile: 201-669-0967 ------------------------------ *From:* michel.thepaut--- via ChimeraX-users <chimerax-users@cgl.ucsf.edu> *Sent:* Wednesday, February 25, 2026 6:15 AM *To:* chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> *Subject:* [chimerax-users] Re: ISOLDE and secondary structure restraints
External Email - Use Caution
Dear Tristan, I have a similar question then I exhume that topic...
As I'm dealing with a ~1500 residues protein that I want to make dance in a density. Then as it is large I would like to use command lines to assign secondary structure to known helices and B-sheets instead of clicking on each residues to say this is an helix and this is a sheet. Is there a way to say to Isolde something like: 20 to 30 is an helix, 50 to 56 is a parallel sheet, ..., 1452 to 1460 is and helix. Please place your tensors and angles and lets dance?...
Thank you for your answer. Michel _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://nam02.safelinks.protection.outlook.com/?url=https%3A%2F%2Fmail.cgl.u... <https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/> _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
-- Altos Labs UK Limited | England | Company reg 13484917 Registered address: 3rd Floor 1 Ashley Road, Altrincham, Cheshire, United Kingdom, WA14 2DT

Not the hard-coded helix and beta-sheet restraints, I’m afraid. But you can get a very similar (probably better, TBH) result with something like: isolde restrain distances #1&(helix|strand)&backbone isolde restrain torsions #1&(helix|strand) sidechain false The advantage of those is that (a) they’ll work a bit better when the true conformation isn’t *perfect* helix or strand, and (b) they have a lot more flexibility in terms of your ability to fine-tune their behaviour (for models fetched from the Alphafold DB, try adding the argument “adjustForConfidence true” to automatically take the pLDDT and PAE values into account). — Tristan On 13 Mar 2022, at 14:42, Guido Hansen via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hi all, I just started model building in cryoEM maps and get great results using ISOLDE on AlphaFold2-derived initial models. I have a question which is mainly directed to Tristan I guess: Is is possible to use the command line/script to apply secondary structure restraints in ISOLDE? I would like to restrain the initial secondary structure assignment from Alphafold especially at resolution worse than 3.5 A. At the moment I select each and every residue range assigned to helix, parallel or antiparallel beta-strand one after the other and turn on the corresponding restrains in the ISOLDE GUI. Is there a better way via command line of script? Cheers, Guido -- PD Dr. Guido Hansen Group Leader [Uni-Lübeck] Universität zu Lübeck Institut für Biochemie Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de<mailto:hansen@biochem.uni-luebeck.de> www.biochem.uni-luebeck.de<http://www.biochem.uni-luebeck.de> Ratzeburger Allee 160 23562 Lübeck _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu<mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Thanks for your suggestions. However, I think that when applying distance restraints using the command below also distances of mainchain atoms of secondary structure elements to nearby sidechain atoms get restrainted. isolde restrain distances #1&(helix|strand)&backbone What you get when using the gui is something like that: -- Guido Am 13.03.2022 um 16:07 schrieb Tristan Croll:
Not the hard-coded helix and beta-sheet restraints, I’m afraid. But you can get a very similar (probably better, TBH) result with something like:
isolde restrain distances #1&(helix|strand)&backbone isolde restrain torsions #1&(helix|strand) sidechain false
The advantage of those is that (a) they’ll work a bit better when the true conformation isn’t *perfect* helix or strand, and (b) they have a lot more flexibility in terms of your ability to fine-tune their behaviour (for models fetched from the Alphafold DB, try adding the argument “adjustForConfidence true” to automatically take the pLDDT and PAE values into account).
— Tristan
On 13 Mar 2022, at 14:42, Guido Hansen via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi all,
I just started model building in cryoEM maps and get great results using ISOLDE on AlphaFold2-derived initial models. I have a question which is mainly directed to Tristan I guess:
Is is possible to use the command line/script to apply secondary structure restraints in ISOLDE? I would like to restrain the initial secondary structure assignment from Alphafold especially at resolution worse than 3.5 A. At the moment I select each and every residue range assigned to helix, parallel or antiparallel beta-strand one after the other and turn on the corresponding restrains in the ISOLDE GUI. Is there a better way via command line of script?
Cheers, Guido
--
*PD Dr. Guido Hansen *Group Leader
Uni-Lübeck
*Universität zu Lübeck* *Institut für Biochemie*
Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de www.biochem.uni-luebeck.de
Ratzeburger Allee 160 23562 Lübeck
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users -- Re: email signature *PD Dr. Guido Hansen* Group Leader
Uni-Lübeck *Universität zu Lübeck* *Institut für Biochemie* Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de www.biochem.uni-luebeck.de Ratzeburger Allee 160 23562 Lübeck
{kind=link}
{kind=link}
Oops! My bad - was responding on the move, and forgetting my own implementation. You're right, the "isolde restrain distances" command works on a per-residue basis, so sidechain atoms will be included regardless. On the other hand, the "isolde release distances" command works per-atom, so after the restrain command you can selectively release the sidechain restraints with: isolde release distances #1&sideonly ________________________________ From: Guido Hansen <hansen@biochem.uni-luebeck.de> Sent: 15 March 2022 16:20 To: Tristan Croll <tic20@cam.ac.uk> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] ISOLDE and secondary structure restraints Thanks for your suggestions. However, I think that when applying distance restraints using the command below also distances of mainchain atoms of secondary structure elements to nearby sidechain atoms get restrainted. isolde restrain distances #1&(helix|strand)&backbone [cid:part1.09p0uiHA.41fj6uxZ@biochem.uni-luebeck.de] What you get when using the gui is something like that: [cid:part2.0mNP8JTc.JBo6Z60y@biochem.uni-luebeck.de] -- Guido Am 13.03.2022 um 16:07 schrieb Tristan Croll: Not the hard-coded helix and beta-sheet restraints, I’m afraid. But you can get a very similar (probably better, TBH) result with something like: isolde restrain distances #1&(helix|strand)&backbone isolde restrain torsions #1&(helix|strand) sidechain false The advantage of those is that (a) they’ll work a bit better when the true conformation isn’t *perfect* helix or strand, and (b) they have a lot more flexibility in terms of your ability to fine-tune their behaviour (for models fetched from the Alphafold DB, try adding the argument “adjustForConfidence true” to automatically take the pLDDT and PAE values into account). — Tristan On 13 Mar 2022, at 14:42, Guido Hansen via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hi all, I just started model building in cryoEM maps and get great results using ISOLDE on AlphaFold2-derived initial models. I have a question which is mainly directed to Tristan I guess: Is is possible to use the command line/script to apply secondary structure restraints in ISOLDE? I would like to restrain the initial secondary structure assignment from Alphafold especially at resolution worse than 3.5 A. At the moment I select each and every residue range assigned to helix, parallel or antiparallel beta-strand one after the other and turn on the corresponding restrains in the ISOLDE GUI. Is there a better way via command line of script? Cheers, Guido -- PD Dr. Guido Hansen Group Leader [Uni-Lübeck] Universität zu Lübeck Institut für Biochemie Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de<mailto:hansen@biochem.uni-luebeck.de> www.biochem.uni-luebeck.de<http://www.biochem.uni-luebeck.de> Ratzeburger Allee 160 23562 Lübeck _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu<mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users -- PD Dr. Guido Hansen Group Leader [Uni-Lübeck] Universität zu Lübeck Institut für Biochemie Tel +49 451 3101 3122 Fax +49 451 3101 3104 E-Mail hansen@biochem.uni-luebeck.de<mailto:hansen@biochem.uni-luebeck.de> www.biochem.uni-luebeck.de<http://www.biochem.uni-luebeck.de> Ratzeburger Allee 160 23562 Lübeck
{kind=link}
{kind=link}
participants (6)
-
 Dan Richman
Dan Richman -
 Guido Hansen
Guido Hansen -
 michel.thepaut@ibs.fr
michel.thepaut@ibs.fr -
 Tristan Croll
Tristan Croll -
 Tristan Croll
Tristan Croll -
 Vorländer,Matthias Kopano
Vorländer,Matthias Kopano